6 years. Has your doctor and stroke hospital done one damn thing with this to get you to 100% recovery? NO? THEN YOU ARE IN INCOMPETENT HANDS, FIRE THEM!
Enhancing Nervous System Recovery through Neurobiologics, Neural Interface Training, and Neurorehabilitation
 Max O. Krucoff1*,
Max O. Krucoff1*,  Shervin Rahimpour1,
Shervin Rahimpour1,  Marc W. Slutzky2,3, V. Reggie Edgerton4 and
Marc W. Slutzky2,3, V. Reggie Edgerton4 and  Dennis A. Turner1,5,6
Dennis A. Turner1,5,6- 1Department of Neurosurgery, Duke University Medical Center, Durham, NC, USA
- 2Department of Physiology, Feinberg School of Medicine, Northwestern University, Chicago, IL, USA
- 3Department of Neurology, Feinberg School of Medicine, Northwestern University, Chicago, IL, USA
- 4Department of Integrative Biology and Physiology, University of California, Los Angeles, Los Angeles, CA, USA
- 5Department of Neurobiology, Duke University Medical Center, Durham, NC, USA
- 6Research and Surgery Services, Durham Veterans Affairs Medical Center, Durham, NC, USA
After an initial period of recovery, human neurological injury has long been thought to be static. In order to improve quality of life for those suffering from stroke, spinal cord injury, or traumatic brain injury, researchers have been working to restore the nervous system and reduce neurological deficits through a number of mechanisms. For example, neurobiologists have been identifying and manipulating components of the intra- and extracellular milieu to alter the regenerative potential of neurons, neuro-engineers have been producing brain-machine and neural interfaces that circumvent lesions to restore functionality, and neurorehabilitation experts have been developing new ways to revitalize the nervous system even in chronic disease. While each of these areas holds promise, their individual paths to clinical relevance remain difficult. Nonetheless, these methods are now able to synergistically enhance recovery of native motor function to levels which were previously believed to be impossible. Furthermore, such recovery can even persist after training, and for the first time there is evidence of functional axonal regrowth and rewiring in the central nervous system of animal models. To attain this type of regeneration, rehabilitation paradigms that pair cortically-based intent with activation of affected circuits and positive neurofeedback appear to be required—a phenomenon which raises new and far reaching questions about the underlying relationship between conscious action and neural repair. For this reason, we argue that multi-modal therapy will be necessary to facilitate a truly robust recovery, and that the success of investigational microscopic techniques may depend on their integration into macroscopic frameworks that include task-based neurorehabilitation. We further identify critical components of future neural repair strategies and explore the most updated knowledge, progress, and challenges in the fields of cellular neuronal repair, neural interfacing, and neurorehabilitation, all with the goal of better understanding neurological injury and how to improve recovery.
Introduction
Historically, for patients suffering from spinal cord injury (SCI), stroke, or traumatic brain injury (TBI), the prognosis for recovery has been poor, and patients with more complete and chronic injuries have shown the least potential for improvement (Jennett et al., 1976; Waters et al., 1992, 1996; Curt et al., 2008; Perel et al., 2008; Steyerberg et al., 2008; Lloyd-Jones et al., 2010). Researchers have been dedicated to improving the quality of life for these patients in several ways, e.g., (1) biological manipulation of the cellular milieu to encourage neuronal repair and regeneration (Magavi et al., 2000; Chen et al., 2002; Lee et al., 2004; Freund et al., 2006; Benowitz and Yin, 2007; Park et al., 2008; Maier et al., 2009; de Lima et al., 2012b; Dachir et al., 2014; Li et al., 2015; Omura et al., 2015), (2) creation of neural- or brain-machine interfaces designed to circumvent lesions and restore functionality (Wolpaw and McFarland, 1994; Kennedy and Bakay, 1998; Leuthardt et al., 2004; Monfils et al., 2004; Hochberg et al., 2006, 2012; Moritz et al., 2008; O'Doherty et al., 2009; Ethier et al., 2012; Collinger et al., 2013; Guggenmos et al., 2013; Ifft et al., 2013; Memberg et al., 2014; Zimmermann and Jackson, 2014; Grahn et al., 2015; Jarosiewicz et al., 2015; Soekadar et al., 2015; Bouton et al., 2016; Capogrosso et al., 2016; Donati et al., 2016; Hotson et al., 2016; Rajangam et al., 2016; Vansteensel et al., 2016), and (3) new rehabilitation techniques that include electrical stimulation and pharmacological enhancement of spinal circuitry to stimulate recovery (Carhart et al., 2004; Levy et al., 2008, 2016; Dy et al., 2010; Harkema et al., 2011, 2012; Dominici et al., 2012; van den Brand et al., 2012; Gad et al., 2013b, 2015; Angeli et al., 2014; Gharabaghi et al., 2014a,c; Wahl et al., 2014; Gerasimenko et al., 2015b). Unfortunately, the path to clinical relevance for these individual approaches remains long, and each field tends to operate largely in its own sphere of influence. Nonetheless, there is now emerging evidence that these methods may synergistically enhance recovery of native motor function that can persist even after the training period and is beyond what was previously thought possible (van den Brand et al., 2012; Guggenmos et al., 2013; Angeli et al., 2014; Wahl et al., 2014; Gad et al., 2015; García-Alías et al., 2015). Some animal models are even displaying functional axonal regrowth, sprouting, and rewiring never seen before in the central nervous system (CNS) of mammals (Bregman et al., 1995; Chen et al., 2002; Liebscher et al., 2005; Freund et al., 2006; Maier et al., 2009; van den Brand et al., 2012; Wahl et al., 2014; García-Alías et al., 2015). Throughout much of this work, evidence is emerging that combinatorial therapy across fields may actually be necessary to achieve significant and lasting neurological repair (Wahl et al., 2014; Gad et al., 2015). This paper explores the state of the art in each of these disciplines, identifies essential components of rehabilitation strategies, and argues why synthesizing approaches across specialties will be essential to realizing clinical applicability.
The Biology of Neurological Injury
In order to understand how best to reverse or repair neurological injury, the mechanisms of cellular development and damage response must be appreciated. During maturation, young neurons of the CNS require activity (stimulated by purposeful actions like vision, walking, or hand function) and trophic factors to survive, grow, and prune (reviewed in Liu et al., 2011). Once mature, however, their axonal growth potential declines due to changes in intrinsic and extrinsic signaling factors, as well as established and stable synaptic fields. After lesioning, the distal portion of an axon undergoes Wallerian degeneration while the proximal portion seals the damaged membrane to form an end bulb (Schlaepfer and Bunge, 1973; Li and Raisman, 1995; Shetty and Turner, 1999; Hill et al., 2001; Fishman and Bittner, 2003). Eventually a growth cone is formed and injured corticospinal axons make an attempt to regrow; however, guidance cues are typically missing, and such efforts are therefore transient, abortive, and ultimately fail (Bernstein and Stelzner, 1983; Magavi et al., 2000; reviewed in Bulinski et al., 1998; Benowitz and Yin, 2007). This inability to regenerate is why injury to the CNS is so devastating and has been considered static once chronic.
After an insult, damaged CNS neurons continue a downward spiral of degeneration known as secondary injury. This includes an uncontrolled release of glutamate from presynaptic vesicles, loss of cell membrane potential, and damage to N-methyl-D-aspartate (NMDA) and a-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors, all of which lead to overstimulation and increased neuronal death (Park et al., 1989; Goforth et al., 1999; Yurkewicz et al., 2005). Nitric oxide, triggered by the activation of NMDA receptors and intracellular calcium, interacts with stress-induced reactive oxygen species to cause DNA fragmentation, lipid peroxidation, and cellular death (reviewed in DeFina et al., 2009; Demirtas-Tatlidede et al., 2012; Villamar et al., 2012). Hemorrhage itself also exacerbates injury, as blood outside vessels releases excitatory amino acids, iron, and thrombin which induce further oxidative stress (Xi et al., 2006). The accumulation of excess intracellular zinc has too been shown to play a role in secondary injury by triggering neuronal death through intrinsic mechanisms such as 5′-adenosine monophosphate-activated protein kinase (AMPK) (Suh et al., 2000; Eom et al., 2016). These mechanisms of primary and secondary neurologic injury are summarized in Table 1.
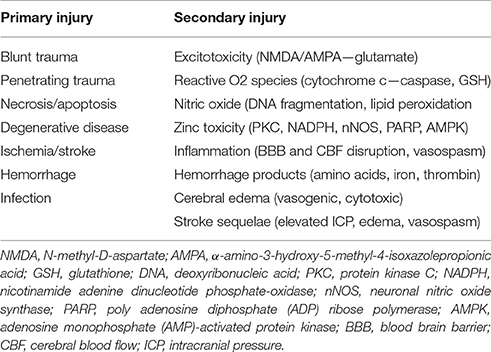
Table 1. Mechanisms of neurological injury.
Upon cell dissolution and fragmentation, inflammation is triggered, resulting in the clean-up of dead cells, disconnection of nonfunctional synapses, release of pro- and anti-inflammatory cytokines, and disruption of the blood-brain-barrier (BBB) (reviewed in Greve and Zink, 2009). This drives astrocytes and endothelial cells to produce more inflammatory mediators and impairs the brain's ability to manage its own perfusion status [i.e., autoregulate cerebral blood flow (CBF)]. During the first hours after injury, decreased perfusion and cerebral ischemia is seen, followed by a second phase of increased perfusion with increased intracranial pressure (ICP), and a final phase of vasospasm and again reduced perfusion (reviewed in Villamar et al., 2012). Until about 10–14 days after injury, inflammation helps to prime the extracellular milieu for subsequent axonal entry and re-innervation (Shetty and Turner, 1995). Resolution of acute inflammation is mediated by apoptosis of the inflammatory cells and endogenous anti-inflammatory mediators (reviewed in Villamar et al., 2012). A brief timeline of the injury environment is provided in Figure 1.
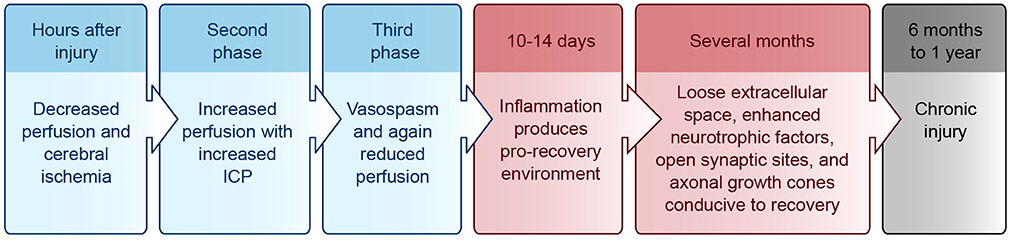
Figure 1. Injury environment timeline. Blue, acute phase; Red, subacute phase; Black, chronic phase. ICP, intracranial pressure.
As a result of these inflammatory processes, cerebral edema tends to emerge during the secondary injury and can be either vasogenic or cytotoxic (Marmarou et al., 2006). Vasogenic edema results from vasodilation, increased permeability of the BBB, and accumulation of molecules in the interstitial fluid. Cytotoxic edema is due to metabolic derangements within cells that lead to changes in osmolality, swelling, and death, often from inadequate metabolism or too severe ionic load for the membrane pumps to handle (reviewed in Greve and Zink, 2009; Zink et al., 2010; Villamar et al., 2012).
While the spinal cord shares certain mechanisms of injury with the brain, its injury patterns also have some important differences. For example, the most frequent primary cause of SCI is traumatic acute compression, and usually at least some subpial neurological tissue is preserved (Wolman, 1965; Tator and Edmonds, 1979; reviewed in Tator, 1995). Mechanical trauma preferentially affects the central gray matter of the cord, likely due to its vascularity and softer consistency, and can cause necrosis, edema, hemorrhage, and vasospasm (Wolman, 1965; reviewed in Tator, 1995). A cascade of secondary pathophysiology similar to that seen in the brain follows, including ischemia, apoptosis, necrosis, fluid and electrolyte disturbances, excitotoxicity, production of free radicals, lipid peroxidation, and an inflammatory response. These processes result in further neurological damage, swelling, and ischemia. Ultimately, a large fluid-filled cavity or cyst forms in the center of the injured cord surrounded by a subpial rim of preserved axons, many of which become demyelinated. Hypertrophic astrocytes and macrophages secrete extracellular matrix and inhibitory molecules that form a glial scar—a physical and chemical barrier to neural regeneration (reviewed in Tator and Fehlings, 1991; Tator, 1995; Mothe and Tator, 2012).
Although intuitively attractive, attempts to mitigate secondary injury and improve recovery with pharmaceutical therapies have been well studied with modest results at best (DeFina et al., 2010; reviewed in Breceda and Dromerick, 2013; Krieger, 2013). In the acute phase of brain injury, suppression of glutamatergic activity appears to be beneficial in minimizing neurological damage and disability (Liu et al., 2013). In the subacute phase, modulation of GABAergic inhibition can minimize the functional impact of an injury (reviewed in Demirtas-Tatlidede et al., 2012). Other drugs that have been used to enhance motor recovery after TBI and/or stroke include naltrexone, bromocriptine, fluoxetine, venlafaxine, levo/carbidopa, donepezil, modafinil, rivastigmine, desipramine, zolpidem, amantadine, methylphenidate, dextroamphetamine, and rasagiline (reviewed in DeFina et al., 2010; Breceda and Dromerick, 2013; Krieger, 2013). Most of these drugs are aimed at normalizing the electrochemical balance of the injured brain to optimize its ability to heal and minimize secondary injury. Dopaminergic medications have also been shown to promote gamma band activity during attention through D4 receptor activation (Kuznetsova and Deth, 2008). Nicotinamide too may help reverse severe oxidation, likely through mitochondrial mechanisms (Shetty et al., 2014), and some authors have suggested that blockade of AMPK in acute brain injury may protect against zinc neurotoxicity (Eom et al., 2016).
Selective serotonin reuptake inhibitors (SSRIs) like fluoxetine play a role in treating depressive symptoms that often accompany neurological disease (thought to be associated with disruption of corticostriatal and thalamocortical loops) (Terroni et al., 2011). Treating depression is crucial to recovery but is often overlooked, as the symptoms of depression and learned helplessness can be found in up to 30% of early stroke patients (Hackett et al., 2014). Vitamins and antioxidants such as essential amino acids, minerals, cofactors, and “immunonutrition” [omega 6 and omega 3 fatty acids, arginine, glutamine, ribonucleic acids (RNAs), mycelia extracts] have all demonstrated modest but generally benign results (DeFina et al., 2010). Additionally, recent evidence suggests that prophylactic anticonvulsants like phenytoin in stroke/TBI are associated with worse functional outcomes, possibly due to reduced axonal/growth cone bursting from sodium channel suppression which may inhibit rewiring (Bhullar et al., 2014). The optimal timing of seizure prophylaxis after brain injury, if beneficial at all, remains open to debate (Thompson et al., 2015).
Molecular Mechanisms of Neural Repair
In addition to the injury mechanisms and glial scar described above, there are many biological mediators that alter the ability of the CNS to repair itself after injury. Intrinsic factors including transcription factors (c-Jun, Atf3, Klf family, Stat3, Sox11, and Smad1) and regeneration-associated genes (Gap43, Cap23, Arg1, Sprr1a, Hspb1, MARCKS, stathmin family, SCG10 L1, P21/waf1, and tubulins) have been shown to alter restoration potential (Grenningloh et al., 2004; Carmichael et al., 2005; reviewed in Sun and He, 2010; Tedeschi, 2011). Phosphatase and tensin homolog (PTEN), a tumor suppressor, seems to play an important role as eliminating its gene has been shown to both prevent apoptosis and induce axon extension in injured retinal ganglion cells (RGCs) (Park et al., 2008; de Lima et al., 2012b). Deletion of Socs3, a suppressor of signaling through the Jak-STAT pathway, also promotes regeneration by enhancing the efficacy of ciliary neurotrophic factor (CNTF) (Smith et al., 2009). If the mechanistic target of rapamycin (mTOR) is inhibited, the regenerative effect of PTEN deficiency is eradicated, suggesting that axon regeneration induced by PTEN deletion is dependent on the mTOR pathway (Park et al., 2008). The proto-oncogene bcl-2 (and expression of its anti-apoptotic protein) also plays a key role in preventing cell death after injury, enabling axonal regrowth in RGCs with the presence of trophic factors and physiologic electrical activity (Chen et al., 1997; Goldberg et al., 2002).
Extrinsic factors that prevent axonal regeneration include inhibitory proteins associated with myelin [e.g., NogoA, myelin-associated glycoprotein (MAG), and oligodendrocyte-myelin glycoprotein (OMgp)], proteoglycans in the perineuronal net and glial scar [e.g., chondroitin sulfate proteoglycans (CSPGs) like aggrecan, versican, brevican, neurocan, NG2, and phosphacan], and molecules that repel axon growth during development which continue to be expressed in the mature CNS (e.g., semaphorins, ephrins, slits, netrins, robos, and Wnts) (reviewed in Benowitz and Yin, 2007; Benowitz and Carmichael, 2010; de Lima et al., 2012a; Omura et al., 2015). A summary of intrinsic and extrinsic factors affecting neural growth and inhibition is provided in Figure 2.
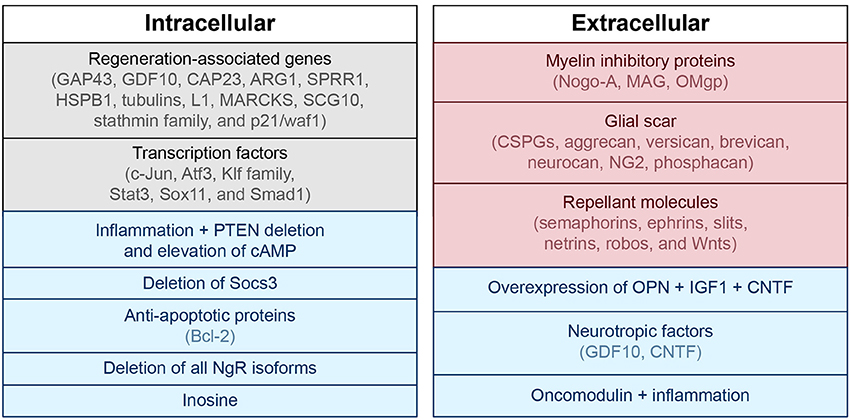
Figure 2. Intra- and extracellular mechanisms of neuronal growth and inhibition. Blue, associated with neuronal growth; Red, associated with neuronal inhibition; Black, modulates both neuronal growth and inhibition. PTEN, phosphatase and tensin homolog; cAMP, cyclic adenosine monophosphate; GAP43, growth associated protein 43; GDF10, growth differentiation factor 10; CAP23, cytoskeleton-associated protein; ARG1, arginase 1; SPRR1, small proline-rich protein 1; HSPB1, heat shock protein family B (small) member 1; MARCKS, myristoylated alanine-rich C-kinase substrate; SCG10, superior cervical ganglion 10; NgR, nogo receptor; CSPG, chondroitin sulfate proteoglycans; NG2, neural/glial antigen 2; MAG, myelin-associated glycoprotein; OMgp, oligodendrocyte-myelin glycoprotein; CNTF, ciliary neurotrophic factor; OPN, osteopontin; IGF1, insulin-like growth factor 1.
Despite identification of these molecules as being important, removal or blockage of extracellular inhibitory factors alone so far has failed to achieve extensive axonal regeneration with a few exceptions (Alilain et al., 2011; reviewed in Benowitz and Yin, 2007; Benowitz and Carmichael, 2010; Liu et al., 2011). Interestingly, a strain of dorsal root ganglion neurons grown from CAST/Ei knockout mice are less inhibited by the same extrinsic cues listed above (Omura et al., 2015). Large regenerative responses have been noted in these cells, and activin seems to play an important role. Also, deletion of receptors that bind to myelin-associated inhibitory molecules (MAIs), or Nogo (NgR) receptors, has been shown to increase regeneration potential in neurons (Dickendesher et al., 2012). This is why anti-Nogo immunotherapies are currently of great interest as potential treatments for neurological injury (Lee et al., 2004; Freund et al., 2006; Maier et al., 2009; Wahl et al., 2014). In fact, immunotherapies aimed at blocking inhibitory factors like NogoA have successfully demonstrated increased sprouting associated with functional recovery in both rat (Bregman et al., 1995; Liebscher et al., 2005; Maier et al., 2009) and primate (Freund et al., 2006) models of SCI. In 2014, Wahl et al. published near full recovery of skilled forelimb function in rats with large strokes after intrathecal injection of an anti-NogoA antibody followed by intensive task-specific training (Wahl et al., 2014). Injection of the NogoA neutralizing agent was shown to promote growth of corticospinal fibers from the intact forebrain motor cortex across the midline of the cervical spinal cord to the hemicord that had lost its input from the motor cortex. This new fiber sprouting was then stabilized by a goal-directed physical therapy regimen. Interestingly, sequential application of drug then training was necessary to show benefit. When immunotherapy and forced-use training were combined simultaneously, functional outcome was poorer compared to no treatment at all or each treatment individually, likely due to abundant but aberrant fiber branching (also seen in Maier et al., 2009). This example outlines the important distinction between regrowth with synapse formation and the restoration of function.
Another example of this principle was demonstrated by Bei et al. in showing that PTEN/SOCS3 co-deletion or overexpression of osteopontin (OPN)/insulin-like growth factor 1 (IGF1)/CNTF could induce regrowth of adult mouse retinal axons to synapse in the superior colliculus, but this connection did not restore visual function on its own (Bei et al., 2016). In fact, these regenerated axons failed to conduct action potentials (APs) due to lack of myelination, and administration of voltage-gated potassium channel blockers was required to enable proper conduction and improve visual acuity.
Despite the litany of inhibitory mechanisms examined above, there are also signals released in the injured brain that are known to promote axonal growth after injury. For example, growth and differentiation factor 10 (GDF10) is induced in stroke and works through transforming growth factor beta receptors I and II (TGFβRI and TGFβRII) to promote axonal outgrowth (Li et al., 2015). Growth associated protein 43 (GAP43), a neuronal growth cone marker, is also induced in peri-infarct cortex after stroke (Stroemer et al., 1995; Schaechter et al., 2006). The purine nucleoside inosine works through a direct intracellular mechanism to induce expression of genes associated with axonal growth (e.g., GAP43, L1, and α-1 tubulin) and has been shown to induce axonal reorganization and improve behavioral outcomes after spinal cord injury and stroke (Zai et al., 2009, 2011; Kim et al., 2013), as well as restore levels of GAP43 in the hippocampus in rats after stroke (Chen et al., 2002; de Lima et al., 2012b; Dachir et al., 2014).
The role of inflammation in axonal regeneration is somewhat controversial. Some components of inflammation cause tissue damage and neuronal death (see The Biology of Neurological Injury section), while others promote cell survival, axon sprouting, and regeneration (Shetty and Turner, 1995; Yin et al., 2003; de Lima et al., 2012a; Kurimoto et al., 2013; Baldwin et al., 2015; reviewed in Benowitz and Popovich, 2011). Both oncomodulin, a macrophage-derived growth factor for RGCs, and injury-induced cytokine release appear to play a role in inflammation-induced axonal regeneration (Yin et al., 2006, 2009; Kurimoto et al., 2013). Traditional anti-inflammatory therapies (e.g., NSAIDs such as ibuprofen) may suppress beneficial as well as deleterious aspects of the immune response, and they can stimulate axonal regeneration via direct effects on neurons (reviewed in Benowitz and Yin, 2008; Benowitz and Carmichael, 2010; Benowitz and Popovich, 2011). When combined with PTEN deletion and elevation of cyclic adenosine monophosphate (cAMP), intraocular inflammation will enable some retinal ganglion cells to regenerate injured axons from the eye to the brain and restore simple visual responses (de Lima et al., 2012b).
The Physiology of Recovery from Neurological Injury
In addition to the molecular and intracellular mechanisms mentioned above, proper function of the neuraxis also relies on the appropriate establishment and maintenance of intercellular mechanisms. Although the CNS does not fully self-repair after injury, neurogenesis does occur naturally in the healthy adult brain. This process happens primarily in the subgranular and subventricular zones (SGZ and SVZ, respectively), and it helps support learning, memory, and olfaction (Doetsch et al., 1999; Laywell et al., 2000; Seri et al., 2001; reviewed in Alvarez-Buylla and Lim, 2004; Ohab and Carmichael, 2008). These areas contain niches of progenitor, glial, and endothelial cells that can self-renew or differentiate into a glial or neuronal lineage. The SGZ supplies the dentate gyrus of the hippocampus and the SVZ gives cells to the olfactory bulb to integrate into local circuitry and support function. There is evidence that damage from stroke stimulates cell proliferation within these zones, and immature neurons are recruited into damaged areas of the striatum and cortex. This process starts at 2 weeks and lasts up to several months after injury (Macas et al., 2006; Thored et al., 2006). Initially, tens of thousands of immature neurons can migrate to damaged areas. However, few mature and survive long-term (Zhang et al., 2001; Arvidsson et al., 2002). While the generation and migration of new neurons to damaged areas is associated with functional recovery, it is possible that behavioral recovery is achieved through mechanisms other than neuronal replacement, e.g., growth factor production in local tissue (reviewed in Ohab and Carmichael, 2008).
Other more prominent mechanisms of restoration of function include reduction in edema, resolution of diaschisis (loss of function in connected areas of the brain due to inactivity, loss of blood flow, and decreased metabolism), and optimization of remaining motor areas (Nudo and Milliken, 1996; reviewed in Feeney and Baron, 1986). Neural plasticity and reorganization occur through the uncovering of previously latent synapses, collateral sprouting of synapses from nearby intact neurons, strengthening or weakening of existing synapses [e.g., through long-term potentiation (LTP) or long-term depression (LTD)], and changes in concentrations of neurotransmitters, ions, gap junctions, and glial cells (reviewed in DeFina et al., 2009; Demirtas-Tatlidede et al., 2012; Villamar et al., 2012; Nahmani and Turrigiano, 2014). These mechanisms are outlined in Table 2. While neural plasticity contributes significantly to functional recovery, it should be noted that not all types are beneficial. For example, maladaptive plasticity and inappropriate axonal sprouting can lead to spasticity, pathological pain, schizophrenia, and seizures (Dimitrijevi and Nathan, 1967; Flor et al., 1995; Teyler et al., 2001; Quartarone et al., 2008; Thickbroom and Mastaglia, 2009; Kuner, 2010; Hasan et al., 2011).
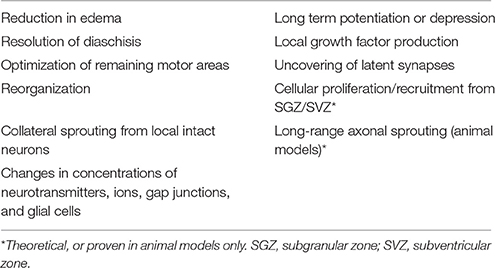
Table 2. Mechanisms of neurological recovery.
Though long-range axonal sprouting was once thought to be non-existent in adult mammals, evidence now supports this possibility in animal models (Chen et al., 2002; Liebscher et al., 2005; Freund et al., 2006; Maier et al., 2009; van den Brand et al., 2012; Wahl et al., 2014). Context-dependent cortical activity and functional growth cones paired with positive feedback seems to be critical for this type of axonal sprouting to generate robust and lasting functional improvement (van den Brand et al., 2012; Wahl et al., 2014). Even without long-range axonal sprouting, some level of functional improvement can occur via other mechanisms of neural plasticity. For example, after stroke, both hemispheres are known to assist with recovery depending on the size of the injury (Dancause, 2005; reviewed in Dancause and Nudo, 2011; Kantak et al., 2012). Following a small stroke within the primary motor cortex (M1) or the corticospinal tract, both ipsilesional dorsal and ventral premotor cortices (PMCs) can reorganize themselves. However, when a lesion involves a larger portion of M1 and the dorsal PMC, the contralesional PMC appears to be critical for recovery-related reorganization (reviewed in Dancause and Nudo, 2011; Kantak et al., 2012). Initiation of post-infarct axonal sprouting from the intact cortical hemisphere to peri-infarct cortex and the contralateral dorsal striatum is signaled by synchronous neuronal activity (Carmichael and Chesselet, 2002). In chronic stroke patients, activity in ipsilesional primary motor and medial-premotor cortices has been shown to be associated with good motor recovery, whereas increased cerebellar vermis activity signals poor recovery (Favre et al., 2014).
For up to several months after the initial injury, the neural environment remains conducive to recovery due to its relatively loose extracellular space, enhanced neurotrophic factors, open synaptic sites, and probing axonal growth cones (Napieralski et al., 1996; Carmichael et al., 2005; reviewed in Nudo, 2013). After 6 months to 1 year, injuries have been classically considered chronic with little opportunity for further gain, although this doctrine is beginning to change (Figure 1) (reviewed in Langhorne et al., 2011; Teasell et al., 2014).
The Neurophysiology Underlying Brain-Machine and Neural Interface Training
Neural- or brain-machine interfaces are electrode-computer constructs that extract and decode information from the nervous system to generate functional outputs. These have been developed to bypass motor lesions (assistive BMIs) (Wolpaw and McFarland, 1994; Kennedy and Bakay, 1998; Leuthardt et al., 2004; Moritz et al., 2008; Ethier et al., 2012; Collinger et al., 2013; Memberg et al., 2014; Jarosiewicz et al., 2015; Bouton et al., 2016; Capogrosso et al., 2016; Hotson et al., 2016; Rajangam et al., 2016; Vansteensel et al., 2016; reviewed in Lobel and Lee, 2014) and, more recently, to facilitate neural plasticity and motor learning to enhance recovery after injury (rehabilitative BMIs) (Carhart et al., 2004; Buch et al., 2008; van den Brand et al., 2012; Ang et al., 2013; Ramos-Murguialday et al., 2013; Wahl et al., 2014; Gharabaghi et al., 2014a,b,c; Gerasimenko et al., 2015b; Donati et al., 2016; reviewed in Ethier et al., 2015; Jackson and Zimmermann, 2012).
Interfaces generally contain at least four components: (1) a method of extracting signals from the nervous system, (2) a way to decode the signals to predict user intent, (3) an output to affect the subject's environment, and (4) a feedback system to help the user refine the output (e.g., visual or other sensory modality). Means of extracting nervous system signals range from invasive [intracortical microelectrodes (APs, or spikes) and larger scale sub- or epidural electrodes (electrocorticography, ECoG)] to non-invasive [electroencephalography (EEG) or electromyography (EMG)]. Targeted outputs have included cursors on a screen (Wolpaw and McFarland, 1994; Kennedy and Bakay, 1998; Leuthardt et al., 2004; McFarland et al., 2010), virtual typing (Jarosiewicz et al., 2015), robotic or prosthetic arms (Collinger et al., 2013; Hotson et al., 2016), wheelchairs (Rajangam et al., 2016), exoskeletons (Donati et al., 2016), the spinal cord (Zimmermann and Jackson, 2014; Capogrosso et al., 2016), and a patient's own extremities (Ethier et al., 2012; Memberg et al., 2014; King et al., 2015; Bouton et al., 2016; Vidaurre et al., 2016).
While initially developed due to the belief that the human nervous system could not self-regenerate (as examined in the previous sections), BMIs have also led to exciting and innovative ways to understand and interact with the nervous system. Current understanding of brain function recognizes an intricate arrangement of interconnected units and circuits that contribute to a larger performing network, as opposed to older models which viewed the brain as a collection of independent anatomical modules with discrete functions (Breakspear and Stam, 2005; Serences and Yantis, 2006; reviewed in Meunier et al., 2010). It has been shown that functionally coupled remote brain locations display near synchronous discharges that represent emergent properties of their assimilated networks (Breakspear and Stam, 2005; Womelsdorf et al., 2007; Stevenson et al., 2012; Menzer et al., 2014). This dispersion of information likely explains why motor information can be found widely distributed throughout the cortex, and how random samples of neurons can provide enough information to reconstruct certain movements in great detail (Carmena et al., 2003; Fitzsimmons et al., 2009; reviewed in Nicolelis and Lebedev, 2009). However, it has also been shown that no matter how well tuned a single neuron is to a behavioral task, that an individual cell only contains limited information and can vary greatly over a short period of time (Wessberg et al., 2000; Carmena et al., 2003). Interestingly, once an ensemble of neurons reaches a certain size, its collective predictive ability plateaus, suggesting that there is redundancy in the neuronal network, and that there are a critical number of neurons required to decode motor intention (Carmena et al., 2003; Vargas-Irwin et al., 2010; reviewed in Donoghue, 2008; Nicolelis and Lebedev, 2009). Collectively, these concepts have led to population algorithms, or decoders that exploit the idea that individual neurons encode multiple parameters with different weights and may vary from trial to trial; however, useful information is maintained among a population instead of individual neurons. The advantage of population decoding systems is that they work even when individual neurons poorly encode motor behavior. These algorithms, in addition to advances in technology that have enabled large-scale recordings of single-neuron activity patterns, have led to the success of many BMIs described in the following section.
The true language of the motor cortex, or how the motor cortex encodes its output signals, is a subject of debate (Vargas-Irwin et al., 2010; Cherian et al., 2011). Coordinated actions of the limbs may engage widespread cortical areas, and M1 is known as the site where motor plans tend to merge before diverging to multiple muscle groups (Vargas-Irwin et al., 2010). While M1 clearly contains kinematic information (joint position and trajectory) sufficient for accurate predictions (Vargas-Irwin et al., 2010), there is evidence that it may more directly encode kinetic (force) variables (Morrow et al., 2007; Cherian et al., 2011; Flint et al., 2014). This would suggest that BMIs built to encode force and/or EMG signals may be more robust across different positional dynamics than trajectory-based BMIs. For gait decoding, there is evidence that motor cortex BMIs may perform better when estimating gait phases or locomotor behaviors as opposed to continuous kinematic variables of leg movement (Rigosa et al., 2015). Interestingly, bimanual arm control appears to have its own representation in the cortex and does not seem to be described simply by a superposition of unilateral movements (Ifft et al., 2013).
As attempts for more dexterous and intuitive control of neural interfaces are pursued, the question of how many independent control signals can be extracted from a neural ensemble arises. The human arm has 7 degrees of freedom (DOF), and the hand has more than 20 (Stockwell, 1981; Jones, 1997). As it turns out, natural grasp postures and reaching-to-grasp movements exist in a much smaller subspace than physically possible movements (Ingram et al., 2008). Dimensionality reduction techniques like principal component analysis (PCA) show that a large proportion of the variance of natural grasping behaviors can be represented by two to three principal components. This provides a strategy for neural interfaces to recapitulate potentially more complex appearing movements while extracting fewer degrees of freedom (reviewed in Hatsopoulos and Donoghue, 2009). An example of such a technique is shown in Figure 3 (Krucoff and Slutzky, 2011).
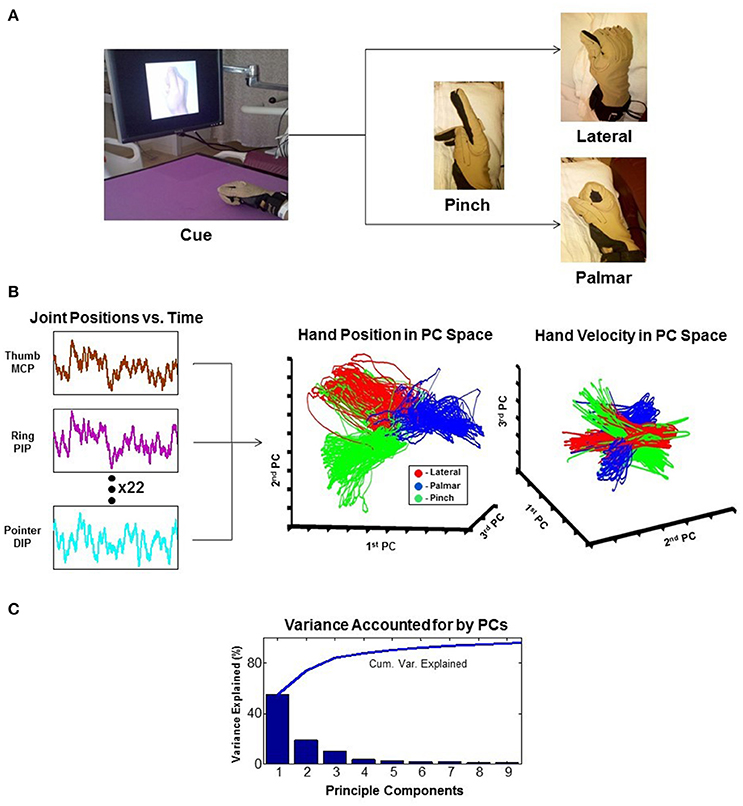
Figure 3. Principal component analysis (PCA). The example reduces 22 joint-position variables of the wrist and fingers to 3 dimensions that represent the state of the hand. (A) Experimental task where the patient is cued to move his hand into one of the three configurations shown. (B) 22 independent variables of hand movement are recorded and reduced to 3 principle components (PCs). Results are plotted as hand position and hand velocity in 3-dimensional PC space. (C) Greater than 80% variance of hand movement is accounted for using only the first three PCs (Krucoff and Slutzky, 2011). MCP, metacarpophalangeal joint; PIP, proximal interphalangeal joint; DIP, distal interphalangeal joint.
In recent years, neural interfaces have been developed to modulate neural plasticity and enhance recovery (rehabilitative BMIs) in addition to bypassing lesions (assistive BMIs). The transition from assistive to rehabilitative BMIs has come with the realization that patients with a chronic neurologic injury may not be at a static level of functioning as previously thought, and that underlying networks even in chronic injury can be modified over time (Guggenmos et al., 2013; reviewed in Jackson and Zimmermann, 2012). A diagram showing the conceptual difference in approach to assistive vs. rehabilitative BMIs is provided in Figure 4. Rehabilitative BMIs pair goal-oriented tasks with expected outcomes and work to activate lesioned circuits to create plasticity for long-lasting improvements. This approach takes advantage of a principle called spike timing dependent plasticity (STDP), or Hebbian plasticity (often expressed as, “neurons that fire together wire together”) (Hebb, 1949; Cooper, 2005). This is the idea that synaptic strength is redistributed to favor functionally relevant pathways that are coincidently active, inferring that both the sign and magnitude of synaptic modification are determined by the precise timing of APs (Rebesco and Miller, 2011). The best known example of Hebbian plasticity is perhaps LTP and LTD in memory circuits. For modeling of complex, larger scale circuits, the Bienenstock–Cooper–Munro (BCM) model maybe more representative of behavior. This theory incorporates both presynaptic and postsynaptic firing rates (Bienenstock et al., 1982; reviewed in Cooper and Bear, 2012) and applies a sliding threshold for LTP/LTD based on post-synaptic activity as the metric for stabilization.
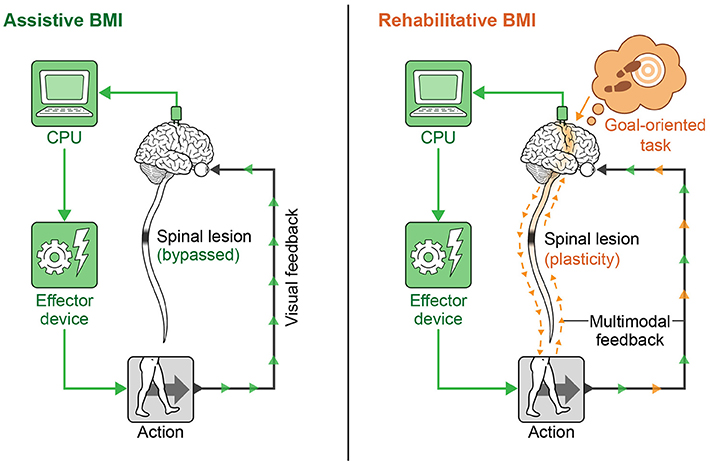
Figure 4. Assistive vs. rehabilitative BMIs. The assistive BMI uses brain signals to bypass a neural lesion and generate an intended action. The rehabilitative BMI pairs goal-oriented tasks with positive feedback and works to re-activate lesioned circuits to create plasticity for long-lasting functional improvement.
A related concept employed in some rehabilitative BMIs is paired associative stimulation (PAS), or the act of pairing stimulation sites to promote plasticity (Stefan et al., 2000; Carson and Kennedy, 2013). An example of a commonly used central stimulation strategy is transcranial magnetic stimulation (TMS). TMS involves applying rapidly changing magnetic fields to the scalp via a magnetic stimulator. Continuous low frequency repetitive stimuli (≤1 Hz rTMS) decreases excitability of targets areas (similar to LTD which is maximally evoked at 1 Hz), while bursts of intermittent high frequency stimuli (≥5 Hz rTMS) enhance excitability (similar to LTP with high frequency bursts) (Kobayashi and Pascual-Leone, 2003; Demirtas-Tatlidede et al., 2012; Shin et al., 2014). These techniques have been used to induce modulation across cortico-subcortical and cortico-cortical networks through trans-synaptic spread, resulting in distant but specific changes along functional networks. Evidence suggests long term effects from TMS may be related to modulation of NMDA glutamatergic receptors, similar to induction of LTP/LTD (reviewed in Villamar et al., 2012). If timed correctly, corresponding sensory inputs can be potentiated (Stefan et al., 2000). Functional electrical stimulation (FES) of paralyzed muscles or electrical stimulation of the nervous system distal to the injury timed with voluntary effort has been shown to accelerate recovery in both SCI and stroke (Daly et al., 2006; Jung et al., 2009; Popovic et al., 2011; Kafri and Laufer, 2015).
mORE AT LINK.
No comments:
Post a Comment