Use the labels in the right column to find what you want. Or you can go thru them one by one, there are only 32,690 posts. Searching is done in the search box in upper left corner. I blog on anything to do with stroke. DO NOT DO ANYTHING SUGGESTED HERE AS I AM NOT MEDICALLY TRAINED, YOUR DOCTOR IS, LISTEN TO THEM. BUT I BET THEY DON'T KNOW HOW TO GET YOU 100% RECOVERED. I DON'T EITHER BUT HAVE PLENTY OF QUESTIONS FOR YOUR DOCTOR TO ANSWER.
Changing stroke rehab and research worldwide now.Time is Brain!trillions and trillions of neuronsthatDIEeach day because there areNOeffective hyperacute therapies besides tPA(only 12% effective). I have 523 posts on hyperacute therapy, enough for researchers to spend decades proving them out. These are my personal ideas and blog on stroke rehabilitation and stroke research. Do not attempt any of these without checking with your medical provider. Unless you join me in agitating, when you need these therapies they won't be there.
What this blog is for:
My blog is not to help survivors recover, it is to have the 10 million yearly stroke survivors light fires underneath their doctors, stroke hospitals and stroke researchers to get stroke solved. 100% recovery. The stroke medical world is completely failing at that goal, they don't even have it as a goal. Shortly after getting out of the hospital and getting NO information on the process or protocols of stroke rehabilitation and recovery I started searching on the internet and found that no other survivor received useful information. This is an attempt to cover all stroke rehabilitation information that should be readily available to survivors so they can talk with informed knowledge to their medical staff. It lays out what needs to be done to get stroke survivors closer to 100% recovery. It's quite disgusting that this information is not available from every stroke association and doctors group.
Assessments
are completely worthless unless they point directly to the 100%
recovery protocols. I see nothing here that suggests you go from the
assessment to the chosen 100% recovery protocol. When the hell will the
stroke medical world do ANYTHING TO GET STROKE SOLVED? I'd have you all
fired! A lot of dead wood needs to removed in stroke and until that
occurs stroke will never be solved! The only use of the word protocol in here is for testing; NOT RECOVERY! I'd have the faculty supervising this fired!
Here's a vicious circle your competent? doctor has to navigate. The best solution would be 100% recovery and getting back to your daily life, but that won't occur, your doctor has been incompetent in getting research done for 100% recovery.
Constipation is commonly seen among patients with
cardiovascular diseases and is linked to adverse outcomes. However, the
association between constipation and the risk of stroke remains
conflicting. Therefore, we aim to conduct a systematic review and
meta-analysis to summarize the available data on this topic.
Methods:
We identified potentially eligible studies from the MEDLINE
and EMBASE databases, searching from inception to May 2024, to
investigate the association between constipation and stroke. To be
included, studies needed to compare the incidence of stroke between
groups with and without constipation. Effect size and 95% CIs were
combined using the generic inverse variance method.
Results:
Our meta-analysis included 8 studies that met the
eligibility criteria. There were 5,360,573 participants, with a mean age
of 53.9 years and 69% are males. We found that patients with
constipation have a 41% increased risk of stroke with a pooled risk
ratio of 1.41 (95% CI: 1.13-1.75; P < 0.01, I2
= 99%) compared with those without constipation. Subgroup analysis
revealed that patients with constipation have a 50% increased risk of
ischemic stroke with a pooled risk ratio of 1.50 (95% CI: 1.15-1.96; P < 0.01, I2 = 99%), but no statistical significance was found for mixed-type stroke outcome.
Conclusions:
Our study revealed that constipation is associated with a
higher risk of stroke. These findings could influence future strategies
for cardiovascular disease prevention and management in patients with
chronic constipation
I'm sure your competent? doctor started prescribing champagne for you 6 years ago. NO? So, your incompetent doctor doesn't follow research on dementia?
You're likely to get dementia. What is your doctors' protocol to prevent
that? 1 bottle equals 5 pours so you need a partner for this.
Shouldn't be hard to find. But nothing on how expensive it needs to be.
But then cheap champagne tastes shitty.
Drinking champagne may be associated with significant cardiovascular benefits, according to a new study published in the Canadian Journal of Cardiology.[1]
The
study identified dozens of lifestyle changes that may help lower a
person’s risk of sudden cardiac arrest (SCA). Some of the changes—eating
more fruit, losing weight—were straightforward, but a few of the
research team’s findings were unexpected. Drinking champagne and/or
white wine, for example, was linked to a reduced SCA risk. The same was
also true for spending more time at a computer—though that may tell us
more about education levels than screen time.
These findings all come from a new exposome-wide association study (EWAS) out of China. The study’s authors explored the UK Biobank study,
focusing on data from more than 500,000 patients. They then looked for
associations between SCA and 125 different modifiable lifestyle factors.
“To
our knowledge, all previous studies on the risk factors of SCA were
hypothesis-driven and focused on a limited number of candidate exposure
factors grounded in previous knowledge or theoretical frameworks,” wrote
first author Huihuan Luo, PhD, a researcher with Fudan University in
Shanghai, China, and colleagues. “This might lead to publication bias
distorting summary conclusions, and might increase the likelihood of
false positive findings resulting from inter-related exposures. More
importantly, the hypothesis-driven approach might miss important
exposures or relationships beyond the predefined hypothesis. To tackle
these limitations, a hypothesis-free, data-driven EWAS has emerged as a
robust analytical framework for simultaneously exploring hundreds of
exposures. This data-driven approach does not rely on previous
knowledge, and facilitates the identification of novel or underexplored
associations.”
Luo
et al. identified 56 different variables that appeared to impact a
person’s SCA risk. Making lifestyle changes based on these variables
could potentially prevent up to 63% of SCA cases.
Spending more
time at a computer, drinking champagne/white wine and eating fruit were
all associated with a reduced SCA risk. On the other hand, negative
“fed-up” feelings, greater arm fat mass, a higher BMI, higher systolic
blood pressure and a lower education level were all associated with a
higher SCA risk.
The researchers did note that these findings
must be examined closely. For instance, spending more time at a computer
may not necessarily be helping patients—instead, it is more likely that
individuals who spend more time at a computer are more likely to have
more education in their background.
Even with that
caveat in place, however, it is clear that modifiable lifestyle factors
make a substantial impact on a person’s odds of SCA.
“Our study
identified a wide range of modifiable factors applicable to general
population, and adherence to corresponding interventions could produce
tremendous public health benefits,” the authors wrote. “Despite
potential overlap among the population attribution fractions of
individual risk factors, they might still be instructive because they
might indicate the benefits of the risk factor itself, as well as its
related factors.”
Is your competent? doctor closely following this because of your risk of Parkinsons post stroke? NO? So, you DON'T have a functioning stroke doctor, do you?
Electrical activity dwindles in cells long before movement issues become visible.
It’s an unsettling thought: You could be walking around for 20 years developing Parkinson’s disease and not even know it.
And once symptoms appear, it’s too late for a cure.
What if a therapy that treats the root causes of Parkinson’s, not just the symptoms, could be started earlier?
Researchers
in the School of Medicine at The University of Texas Health Science
Center at San Antonio are studying changes in Parkinson’s-affected cells
at various stages of the disease, long before any symptoms are evident.
They describe the changes in an April issue of the Journal of Neuroscience.
The
hope of the research is twofold: 1) gain understandings that can be
used to formulate a drug to arrest the disease at a halfway point, and
2) lengthen the time when patients with Parkinson’s can lead healthy,
productive lives.
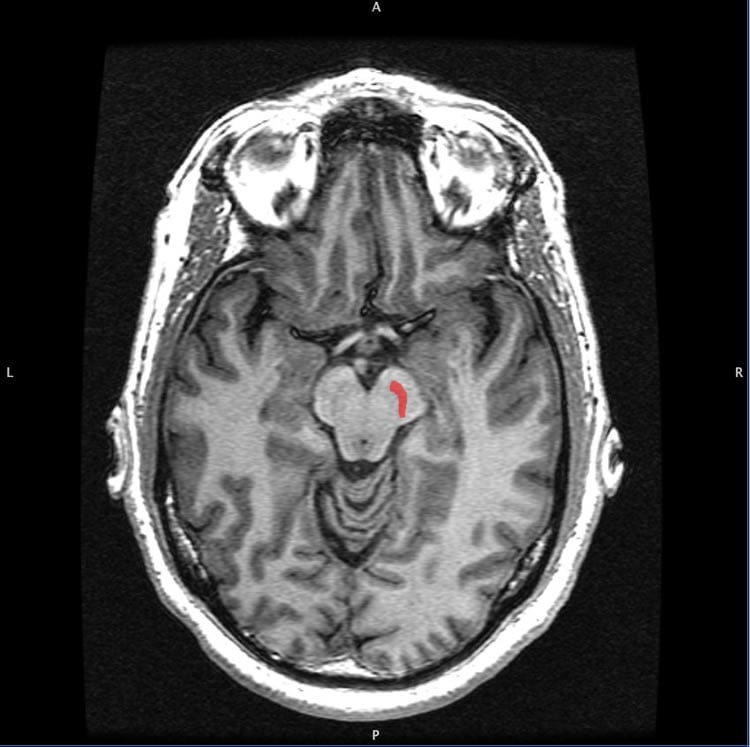
Parkinson’s
is marked by the degeneration and death of cells called dopamine
neurons. These neurons are found in a brain structure called the
substantia nigra. Image is for illustrative purposes only. Credit: Geoff
B Hall.
Hidden changes
“For the first time we are
getting a look at what’s going on in the time window before the disease
visibly takes hold but while changes are occurring,” said study senior
author Michael Beckstead, Ph.D., assistant professor of physiology and a
member of the Barshop Institute for Aging and Longevity Studies at the
UT Health Science Center.
Parkinson’s
is marked by the degeneration and death of cells called dopamine
neurons. These neurons are found in a brain structure called the
substantia nigra. The Health Science Center researchers studied mice in
which only these neurons are affected by a genetic mutation.
The MitoPark mouse, as it is called, is designed so that
mitochondrial activity is hampered just in dopamine neurons of the
substantia nigra. Mitochondria produce energy for our cells, and since
these mice have impaired mitochondria, their dopamine neurons don’t make
energy efficiently.
Mimics human Parkinson’s
At
first, the mice are completely normal, but as weeks and months go by,
the mutation causes their dopamine neurons to slowly become sick and die
off. “It’s a progressive model in that these changes don’t take place
overnight,” Dr. Beckstead said. “This makes it like the human disease,
which is thought to be somewhere in the range of a 20-year process
before symptoms become evident.”
In MitoPark mice, behavioral
symptoms such as tremor start to manifest when the mice are about 20
weeks old. The UT Health Science Center study assessed functional status
at time points before that — comparing dopamine neuron function at 6-10
weeks of age with function at 11-15 weeks of age and function at
16-plus weeks.
Timeline of decline
With
these comparisons, the researchers constructed a timeline of functional
decline in the dopamine neurons. They observed changes in three
categories:
Smaller dopamine neurons
Reduced communication between the neurons
Impaired electrical activity of the neurons
“Pretty
much everything we measured declined in these cells,” Dr. Beckstead
said. “It was really remarkable how everything we studied changed. It
was a general decline, and these changes were all occurring before the
animals were symptomatic, before you could detect any sort of deficit in
their movement.”
In older mice
starting to display the abnormal movements of the disease, the
scientists made another observation — heightened gene expression to
increase the electrical activity in the dopamine neurons.
“This is a late occurrence in the disease process,” Dr. Beckstead
said. “We believe the cells are trying to compensate for the declining
electrical activity. That’s probably how humans are able to be free of
symptoms for so long when they have Parkinson’s, even though 30 percent
or more of their dopamine neurons have died out.”
The study
results aren’t going to translate into a clinical therapy any time soon,
but such findings offer the promise that one day the root cause of
Parkinson’s may be understood and treated.
Current treatments for
Parkinson’s disease are all symptomatic. They focus on improving the
movement deficits and making the patients more comfortable.
“We
don’t have any treatments right now that actually affect the disease
process,” Dr. Beckstead said. “The reason we don’t have any is we don’t
understand what’s going on in the early stages of this disease. Studies
such as ours will help fill in those knowledge gaps.”
About this genetics research
Funding:
Grants from the William and Ella Owens Medical Research Foundation, the
National Institutes of Health and the U.S. Department of Veterans
Affairs supported this work.
Source: Will Sansom – UT Health Science Center San Antonio Image Credit: The image is credited to Geoff B Hall and is in the public domain. Original Research:Abstract
for “Dopaminergic Neurons Exhibit an Age-Dependent Decline in
Electrophysiological Parameters in the MitoPark Mouse Model of
Parkinson’s Disease” by Sarah Y. Branch, Cang Chen, Ramaswamy Sharma,
James D. Lechleiter, Senlin Li, and Michael J. Beckstead in Journal of Neuroscience. Published online April 6 2016 doi:10.1523/JNEUROSCI.1395-15.2016
Nicotinamide
adenine dinucleotide (NAD) augmentation is a multi-target therapeutic
approach that impacts on multiple disease pathways across
neurodegenerative diseases (NDDs).
NAD augmentation shows strong preclinical evidence of benefit in several NDDs.
Early-phase clinical trials show encouraging results regarding target engagement and preliminary efficacy in several NDDs.
Current
data indicate that NAD augmentation therapy is safe, but long-term data
in appropriately sized cohorts are not yet available.
The
number of clinical trials testing NAD augmentation in NDDs is rising
rapidly, but long-term, well-planned, and adequately powered efficacy
trials are urgently needed.
Abstract
Neurodegenerative
diseases (NDDs) pose a significant and rapidly growing global health
challenge, but there are no effective therapies to delay or halt
progression. In recent years augmentation of nicotinamide adenine
dinucleotide (NAD) has emerged as a promising disease-modifying strategy
that targets multiple key disease pathways across multiple NDDs, such
as mitochondrial dysfunction, energy deficits, proteostasis, and
neuroinflammation. Several early clinical trials of NAD augmentation
have been completed, and many more are currently underway, reflecting
the growing optimism and urgency within the field. We discuss the
rationale and evolving therapeutic landscape of NAD augmentation. We
argue that, to fully realize its therapeutic potential, it is essential
to determine the specific contexts in which NAD supplementation is most
effective and to address crucial knowledge gaps.
Keywords
neurodegenerative disease
Parkinson's disease
therapeutic
The challenge of neurodegenerative diseases
Over 80 million people are currently living with a neurodegenerative disease (NDD; see Glossary)
such as Parkinson's disease (PD) or Alzheimer's disease (AD), and this
number is expected to double within the next 20–30 years [1., 2., 3.]. No disease-modifying therapies (DMTs)
are available to halt or prevent disease progression, and affected
individuals face functional decline, early institutionalization, and
significantly shortened life expectancy [4,5].
Consequently, NDDs impose a staggering socioeconomic burden, which has
already surpassed the costs of cancer and cardiovascular disease
combined, and is increasing exponentially [6., 7., 8.].
Although
the etiopathogeneses of NDDs are unknown, they share several
pathological and pathophysiological hallmarks. Neuronal dysfunction and
degeneration occur across multiple regions of the nervous system in a
partly disease-specific but overlapping distribution [9., 10., 11.]. Some form of protein misfolding and aggregation, commonly referred to as neurodegenerative proteinopathy,
occurs across NDDs. This primarily includes α-synuclein in PD, dementia
with Lewy bodies (DLB), and multiple system atrophy (MSA); amyloid-β
and tau in AD; tau in progressive supranuclear palsy (PSP) and
corticobasal degeneration (CBD); and TDP-43 in frontotemporal dementia
(FTD) and amyloid lateral sclerosis (ALS) [11., 12., 13.].
However, mixed-type proteinopathy involving a combination of these
pathologies is an increasingly recognized phenomenon across NDDs [11., 12., 13.]. Although these abnormalities are often attributed to aberrant proteostasis
caused by proteasomal and lysosomal dysfunction, the precise mechanisms
underlying the origin and propagation of the proteinopathy remain
unknown.
Mitochondrial
dysfunction is another well-established pathological hallmark observed
across the spectrum of NDDs, and commonly manifests as quantitative
and/or functional deficiencies in mitochondrial respiratory chain (MRC) complexes, particularly affecting complex I and, to a lesser degree, complex IV [14., 15., 16., 17.].
In addition, impaired mitochondrial DNA (mtDNA) maintenance, reactive
oxygen species (ROS)-mediated cellular stress, and altered mitochondrial
dynamics and quality control have been reported [18., 19., 20.].
Furthermore, neuroinflammation, particularly involving microglial and
astrocyte activation, as well as secretion of proinflammatory cytokines,
is believed to be involved in the pathogenesis of NDDs [21., 22., 23.].
Most past and present disease-modifying strategies for NDDs target one or more of the aforementioned processes [24,25].
However, no therapeutic intervention has yet achieved significant
disease modification in clinical trials, with the notable exception of
mild but encouraging effects of some immunotherapies against amyloid-β
in AD [26,27].
One emerging therapeutic candidate in aging and neurodegeneration
research is NAD, a vital molecule that is essential for all life. As a
crucial redox cofactor in nutrient and energy metabolism pathways, NAD
constantly shuttles between its oxidized (NAD+) and reduced
(NADH) forms. Furthermore, NAD can be phosphorylated to NADP, which
participates in anabolic redox reactions, the immune response, and
oxidative stress defense [28]. In addition, oxidized NAD+
is a substrate for a multitude of signaling reactions, including
protein, DNA, and RNA modifications and the generation of
second-messenger molecules, all of which require hydrolysis of the
molecule, removal of the nicotinamide (Nam) moiety, and transfer of the
remaining ADP-ribose moiety onto an acceptor molecule [29].
These reactions regulate processes vital to cell survival, including
DNA repair via the activity of poly(ADP-ribose) polymerases (PARPs),
epigenetic regulation of gene expression (e.g., via histone
deacetylation by NAD+-dependent sirtuins), and calcium signaling. NAD+-dependent
signaling leads to the degradation of the molecule, and constant NAD
biosynthesis is therefore necessary to replenish NAD levels.
However,
NAD levels have been shown to decline with age, a phenomenon observed
in multiple tissues including the brain in model organisms [30], supported by cross-sectional evidence in humans [31., 32., 33.], although true longitudinal studies over the human age spectrum are still lacking [34].
Although the precise mechanisms underlying age-related NAD decline
remain unknown, it is primarily attributed to the increased activity and
expression of NAD-degrading enzymes such as CD38 – an NAD
glycohydrolase that hydrolyzes NAD and generates second-messenger
molecules [35].
This age-dependent decline of NAD is hypothesized to contribute to the
onset and progression of age-related diseases including
neurodegeneration [36,37].
Conversely, augmenting NAD metabolism via supplementation of precursors
has been shown to confer rejuvenating effects, including lifespan and
healthspan extension in multiple animal models, neuroprotection in
preclinical models of neurodegeneration, as well as promising
early-phase clinical findings [30].
Building on these results, treatment with biosynthetic NAD precursors
has moved into focus as a potential multi-target intervention for NDDs.
In this review we discuss the rationale of NAD supplementation as
therapeutic approach for NDDs and highlight crucial knowledge gaps in
the field.
Clinical NAD augmentation approaches
NAD biosynthesis is multifaceted and can be initiated from several precursors or biosynthetic intermediates (Box 1).
Enhancing NAD biosynthesis via precursor supplementation is currently
the most widely used strategy for augmenting NAD levels in clinical
applications. The most commonly utilized precursors are NR and NMN,
although nicotinic acid (NA) is also being investigated. Unlike NR and
NMN, NA binds to the GPR109A receptor – also referred to as HCAR2 and
now commonly known as niacin receptor 1 (NIACR1). NA exerts a
lipid-modifying effect and was applied in the clinic for decades as a
cholesterol-lowering agent before the introduction of statins [38].
However, NIACR1 activation also triggers skin flushing, an unpleasant
side effect that may limit tolerability. This can be mitigated or
prevented through the use of extended release formulations and/or
gradual dose escalation at the start of treatment [39,40].
Box 1
NAD biosynthesis
NAD
biosynthesis generally takes place in two stages in which nucleosides
or bases are first converted to the mononucleotide, followed by
condensation to a dinucleotide by the addition of AMP from ATP [97,98]. The major pathway in mammals – the salvage pathway – uses nicotinamide (Nam), which is also a degradation product of NAD+-dependent
signaling reactions, to generate nicotinamide mononucleotide (NMN) via
nicotinamide phosphoribosyltransferase (NAMPT, Figure I).
The acid form of Nam, niacin or nicotinic acid (NA), is also the major
entry point to NAD synthesis in bacteria and plants and is converted to
nicotinic acid mononucleotide (NAMN) by nicotinic acid
phosphoribosyltransferase (NAPRT) via the Preiss–Handler pathway.
Recently, trigonelline (Trg), a methylated form of NA, was shown to
support NAD biosynthesis by generating NA via a so far unidentified
process. Quinolinic acid (QA), which is generated by de novo
synthesis from tryptophan (Trp) via the kynurenine pathway, also yields
NAMN through conversion by quinolinic acid phosphoribosyl transferase
(QPRT). All three enzymes require phosphoribosyl pyrophosphate (PRPP) as
a cosubstrate. Nicotinamide riboside (NR) and its acid form nicotinic
acid riboside (NAR) also yield the mononucleotides NMN and NAMN,
respectively, by conversion via nicotinamide riboside kinases (NRKs).
The reduced form of NR, NRH, is a substrate for adenosine kinase (ADK)
and yields the reduced form of NMN, NMNH. The generation of the
dinucleotides is catalyzed by the family of nicotinamide/nicotinic acid
mononucleotide adenylyl transferases, N(A)MNATs, three of which are
present in humans [98,99]. This yields either NAD+ from NMN, NADH from NMNH, or nicotinic acid adenine dinucleotide (NAAD) from NAMN. Conversion of NAAD to NAD+ is carried out by NAD synthetase (NADS or NADSYN1).
The
major NAD precursors used in clinical trials (NR, NMN, and NA) are
highlighted in green. This figure was created with BioRender.
Beyond
precursor supplementation, alternative strategies for NAD augmentation
focus on reducing NAD consumption by inhibiting the activities of
NAD-degrading enzymes such as PARPs and NAD glycohydrolases including
CD38 and SARM1. PARP inhibitors are primarily investigated as cytotoxic
agents in cancer therapy [41],
but are also being explored for their potential to preserve NAD levels.
NAD glycohydrolase inhibition, for example by small-molecule inhibitors
of CD38, has been shown to increase NAD levels and reverse
aging-related NAD decline in mice [42,43], as well as to increase lifespan and healthspan in a mouse model of chronological aging [44].
In addition, targeting the enzymes that regulate the availability of
NAD biosynthetic precursors or intermediates presents another avenue for
intervention. One such enzyme, purine nucleoside phosphorylase (PNP),
has been identified as a major factor for NR degradation in human cell
and mouse experiments [45]. Conversely, activating key NAD biosynthetic enzymes, such as NAMPT, has been shown to elevate cellular NAD levels [46] and has shown promise in a mouse model of diabetes [47].
NAD supplementation as a disease-modifying strategy for NDDs
NAD
augmentation treatment for NDDs is not an entirely new concept. In the
1980s and 90s, intravenous and oral NADH administration was tested in
open-label studies for PD, with reportedly positive effects on
disability and function, and it was hypothesized that this effect may be
mediated by stimulation of dopamine synthesis [48., 49., 50.].
However, the lack of blinded, placebo-controlled trials at the time
made it impossible to determine whether the observed effects were
attributable to NADH or were placebo effects. This concern is
particularly relevant in PD where placebo effects are well-documented
and can significantly influence outcomes [51].
In recent years this field was revitalized with the introduction of
precursors, and the first randomized blinded trials with NR in PD were
reported in 2022 and 2023, marking a significant milestone in the area
of NAD augmentation therapy [52,53].
Alongside these developments, there has been a surge of clinical trials
investigating NAD augmentation strategies in various NDDs, primarily
using NR and NMN, although NA is also being evaluated (Table 1).
Table 1
NAD precursor
Disease/condition
Phase
Dosage (mg/day)
Duration
Outcome(s)
NAD measurements
Clinical trial ID
NR
MCI/AD
1
1000
12 weeks
Brain energy metabolism and mitochondrial function Cognitive function
Brain NAD+ and NAD+/NADH ratio
NCT04430517
AD
2
1000, 2000, 3000
12 weeks
Cerebral and CSF NAD FDG-PET Safety Clinical assessment
Rationale of NAD augmentation as a disease-modifying strategy in NDDs
A
rapidly growing body of preclinical results and emerging clinical
evidence suggests that increasing cellular NAD levels may exert
neuroprotective, disease-modifying effects in NDDs. This premise is
supported by multiple NAD-regulated pathways and mechanisms that
influence key pathophysiological processes implicated in NDDs (Figure 1).
One of the most compelling arguments lies in the observation that NAD
levels decline with age, which remains the strongest known risk factor
for neurodegenerative diseases such as AD and PD. Restoring brain NAD
levels may therefore counteract age-related cellular stress and
vulnerabilities and mitigate neurodegeneration. Moreover, NAD is
essential for mitochondrial function because NADH is the substrate for
MRC complex I. Mitochondrial dysfunction, particularly complex I
deficiency, is a hallmark of many NDDs including AD and PD [20].
Given that mitochondrial metabolism is heavily dependent on NAD
availability, NAD augmentation has the potential to enhance
mitochondrial function and improve cellular bioenergetics. This is
supported by evidence from multiple preclinical models [54], as well as by a recent clinical trial on mitochondrial myopathy where NAD supplementation led to improved clinical outcomes [40]. NAD augmentation also supports increased synthesis of NADP, a central factor in cellular antioxidant defense [28].
Although direct clinical evidence of antioxidant effects by NAD
augmentation is lacking, a recent clinical study demonstrated
significantly elevated NADP levels in blood upon high-dose NR
supplementation [53]. In addition, NAD supplementation reduces neuroinflammation, a key component of NDDs [55] in preclinical models [56], and also reduces inflammation biomarkers in humans [52].
Figure 1. Nicotinamide adenine dinucleotide (NAD) augmentation in neurodegenerative disease (NDD) is a multi-target approach.
NAD
supplementation has the potential to affect several disease-relevant
pathways that overlap in NDDs. These include pathways of protein
homeostasis and turnover such as lysosomal degradation and autophagy,
mitochondrial function and quality control, genetic and epigenetic
transcription regulation and DNA repair mechanisms, oxidative stress
defense, and neuroinflammatory mechanisms. Abbreviations: MRC,
mitochondrial respiratory chain; PARP, poly(ADP-ribose) polymerase. This
figure was created with BioRender.
Furthermore,
NAD is central in DNA repair pathways, for example via the activity of
PARP1. Enhancing NAD levels could therefore confer resilience to the DNA
damage which accumulates in aging and NDDs [57].
Likewise,
epigenetic dysregulation has been implicated in NDDs. For instance,
changes in histone acetylation have been observed in postmortem brain
samples from individuals with PD [58]. Sirtuins, a family of NAD+-dependent
deacylases, play a key role in maintaining epigenetic balance. Reduced
NAD levels may impair sirtuin activity and thus contribute to disease
progression. Finally, NAD augmentation in humans has been shown to
upregulate the expression of genes encoding lysosomal and proteasomal
components, structures that are crucial for maintaining proteostasis and
organellar quality control [52].
Thus, NAD augmentation may act as a multitarget strategy that exerts
synergistic effects across several crucial pathways for NDDs, thereby
enhancing cellular resilience to disease.
Parkinson's disease and related disorders
In
preclinical research, NR was shown to alleviate mitochondrial function
in induced pluripotent stem cell (iPSC)-derived neurons from an
individual with glucocerebrosidase (GBA)-related PD, and to rescue the
phenotype in Drosophila models of the same disorder [54]. In Caenorhabditis elegans, NAD+ treatment protected against methylmercury-induced dopaminergic neurodegeneration and ensuing behavioral deficits [59]. More recently, NR alleviated disease-related phenotypes in a C. elegans
model of α-synuclein overexpression, possibly by activation of the
mitochondrial unfolded protein response, and partially rescued
behavioral dysfunction in a proteasome inhibition model of parkinsonism
in mice [60].
Similarly, NR reduced motor dysfunction, increased survival time, and
attenuated neurodegeneration in a toxic model of
1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced dopaminergic
degeneration in zebrafish [61].
Interestingly, NAD biosynthetic enzymes have also been found to exhibit
moonlighting activity as molecular chaperones that can improve
proteostasis and positively influence α-synuclein misfolding in a yeast
model of PD [62].
Early-phase
clinical trials exploring NAD replenishment in PD have yielded
promising results while raising no significant safety concerns. The
NADPARK study, a Phase 1 randomized double-blind trial, assessed target
penetration and engagement of oral NR (1000 mg/day) in newly diagnosed,
treatment-naïve PD patients. The study showed that oral administration
of NR augments NAD metabolism both systemically and in the central
nervous system (CNS), and this is associated with altered brain
metabolism and mild clinical improvement. Furthermore, NR led to reduced
inflammatory cytokines in cerebrospinal fluid (CSF) and changes in gene
expression linked to mitochondrial function, antioxidant activity, and
protein degradation pathways in peripheral tissues [52]. Another small trial using 1H magnetic resonance spectroscopy (MRS) provided further support that NR supplementation increases cerebral NAD levels [63].
Building
on this foundation, the NR-SAFE trial evaluated the safety and
tolerability of a higher NR dose (3000 mg/day); it was well tolerated
for a period of 30 days and led to robust NAD metabolome augmentation in
blood, including increases in NAD+, the NAD+/NADH ratio, and NADP+.
Interestingly, a potential symptomatic benefit was also observed.
Although a transient rise in serum homocysteine was noted, it remained
within the normal range and did not raise safety concerns [53].
Several
larger-scale and ongoing studies are now expanding on these findings.
The N-DOSE study is a Phase 2 dose-escalation trial designed to identify
the optimal biological dose of NR for PD, with completion anticipated
in early 2025 (NCT05589766). The NOPARK study is a randomized,
double-blind, Phase 3 trial which aims to evaluate whether NR can slow
clinical disease progression in early-stage PD. With 400 participants
enrolled, the trial is expected to conclude by mid-2025 (NCT03568968).
An open-label extension study, NOPARK-extension, will further assess the
long-term safety and tolerability of NR (1000 mg/day) for a period of
up to 4 years (NCT05546567). The NADAPT study is a Phase 2 randomized,
double-blind trial with the primary objective to evaluate whether
high-dose NR (3000/day) can delay the progression of atypical
parkinsonism syndromes, including progressive supranuclear palsy (PSP),
multiple system atrophy (MSA), and corticobasal syndrome (CBS;
NCT06162013).
Evidence in Alzheimer's disease
In a C. elegans
model of AD, NMN supplementation restored defective mitophagy, a
phenotype also observed in human brains with AD, and attenuated
cognitive decline [64]. NR has shown multiple beneficial effects in C. elegans
and mouse models of AD, including increased mitochondrial proteostasis
and mitochondrial gene expression, as well as reduced amyloid-β
proteotoxicity, β-secretase levels, neuroinflammation, and cellular
senescence, and also restored cognition [65., 66., 67.].
In a DNA repair-deficient AD model, NR reduced tau pathology and
neuroinflammation, and improved synaptic transmission and cognition [68].
Similarly, supplementation with Nam was shown to reduce phosphorylated
tau levels and to increase the acetylation levels of sirtuin substrates [69].
Early
clinical trials with NAD precursors in AD or related conditions have
provided mixed results so far. A study in healthy adults supplemented
with NR (1000 mg/day for 6 weeks) reported augmented NAD levels in
plasma extracellular vesicles enriched for neuronal origin (NEVs), and
this was accompanied by reduced levels of amyloid-β42 (Aβ42) and
neuroinflammatory markers [70].
However, a study in older adults with mild cognitive impairment (MCI)
receiving NR 1000 mg/day or placebo for 10 weeks showed no change in
cognition or other disease-related outcome measures [71].
Furthermore, a recent Phase 2 study of high-dose Nam (3000 mg/day) in
persons with AD reported no significant changes in CSF levels of total
tau, phosphorylated tau, or amyloid-β. Although a nominally significant
effect was observed on cognitive decline, this did not survive multiple
testing correction [72]. Several studies with NAD precursors in AD or MCI are currently ongoing (Table 1).
Our N-DOSE_AD trial is exploring the optimal dose of NR in AD and
monitors brain metabolism and cognitive function over a course of 3
months (NCT05617508). Another Phase 1 trial in MCI and mild Alzheimer's
dementia is exploring the effect of 12 weeks NR on brain NAD levels and
cerebral metabolism, as well as clinical and cognitive outcomes
(NCT04430517). Moreover, a proof-of-concept trial is testing NMN
treatment for 90 days with the aim of increasing sirtuin activity in the
brain of individuals with AD and is monitoring among others circulating
biomarkers of aging (NCT05040321). Treatment with NA is also being
explored, although this focuses less on NAD augmentation and more on the
ability of NA to bind to the receptor HCAR2 (i.e., NIACR1) and a
potential related microglia response (NCT06582706).
Evidence in ALS and other NDDs
NAD
augmentation has shown promising preclinical evidence for ALS,
including reduced astrocyte-mediated neurotoxicity via SIRT6 activation
in cells isolated from mice overexpressing mutant human SOD1 [73], attenuated mitochondrial dysfunction and improved function in motor neurons of a TDP-43-overexpressing mouse model [74], and delayed progression of motor impairment in mice carrying Sod1 mutations [75,76].
Moreover, the expression of several NAD biosynthesis enzymes and SIRT6
was decreased in postmortem spinal cord samples of persons with ALS,
indicating that these pathways may be impaired [76].
An early, small-scale randomized double-blind clinical trial assessing
the safety and efficacy of NR in combination with pterostilbene in ALS
reported a significant clinical improvement in the treatment group [77]. The efficacy of this treatment is currently being assessed in the ongoing Phase 2 NO-ALS trial (NCT04562831, Table 1).
NAD
augmentation has shown promising preclinical and/or clinical effects in
several other neurodegenerative diseases. A few selected examples are
mentioned in the following text. Encouraging preclinical effects have
been reported in models of Huntington's disease [78], possibly mediated via the sirtuin1–PGC-1α–PPAR pathway [79]. A first clinical trial of NAD supplementation in Huntington's disease (NAD-HD) is planned to start in 2025 [80].
Furthermore, NAD treatment has shown beneficial preclinical effects in
worm and mouse models of ataxia telangiectasia (AT), including enhanced
lifespan and healthspan, likely via supporting mitophagy and DNA repair [81].
Two recently completed open-label trials of NR treatment in AT have
reported highly encouraging results, including clinical attenuation of
ataxia and amelioration of eye movements [82,83]. Another trial in AT is ongoing (NCT06324877, Table 1). In multiple sclerosis the importance of NAD has long been recognized [84],
and preclinical studies have indicate beneficial effects of restoring
NAD levels on demyelination and axonal damage, for instance via
inhibiting the NLRP3 inflammasome [85]. A first clinical trial of NR supplementation in multiple sclerosis is ongoing (NCT05740722, Table 1).
Safety and risks of high-dose NAD supplementation and alternative strategies for NAD augmentation
According
to the National Institutes of Health (NIH) Office of Dietary
Supplements, the recommended daily vitamin B3 intake is 14 mg niacin
equivalents (NE) for women and 16 mg NE for men. Thus, the doses
currently tested in clinical trials – typically 1000–3000 mg/day – by
far exceed the daily requirements for healthy, middle-aged persons.
Nevertheless, no major adverse events have been reported so far. NA has
been used in doses up to 2000 mg/day as an antilipidemic and, although
transient skin flushing and gastrointestinal side effects were common,
severe adverse events were rarely seen [86]. Moreover, skin flushing and gastrointestinal symptoms can be effectively mitigated by slow dose titration.
NR and NMN have so far demonstrated an excellent safety profile at up to 3000 mg and 1250 mg, respectively [53,87].
However, most trials to date have been very short, generally <6
months, and data from larger and longer studies are lacking. For NR,
this will hopefully be addressed by the NOPARK and NOPARK-extension
trials which will assess safety for up to 4 years.
NAD augmentation leads to elevated levels of excretion products such as methyl-Nam and methylated pyridones N-methyl-6-pyridone-3-carboxamide and N-methyl-4-pyridone-5-carboxamide
(N-Me-6-PY and N-Me-4-PY, also known as 2PY and 4PY, respectively). The
synthesis of these compounds requires methylation of Nam using S-adenosyl
methionine (SAM) as the methyl donor. It has been speculated that
increased utilization of SAM could lead to reduced methyl group
availability, thus affecting other important pathways such as DNA
methylation and leading to a harmful increase in homocysteine, a
downstream intermediate of SAM utilization. However, thus far, all
indications point to only a mild effect on homocysteine levels and no
clinically relevant impact on systemic methylation metabolism [53,88]. Data from longer interventions will be necessary to provide conclusive evidence on this matter.
A
theoretical risk of prolonged NAD augmentation is growth facilitation
for existing cancers. This is supported by a recent study in mice that
showed both increased tumor growth and metastasis formation on an
NR-rich diet [89].
In fact, inhibition of NAD biosynthesis (e.g., using NAMPT inhibitors)
and interference with NAD-dependent pathways (e.g., DNA repair by PARP1)
are emerging strategies for cancer treatment. In contrast to
facilitating the growth of existing tumors, no convincing data have been
reported to date supporting a carcinogenic or genotoxic effect of NAD
supplementation. Moreover, the safety profile of NAD precursor
supplementation (in this case, NR) revealed no carcinogenic effect at
3000 mg/kg/day in rats [90], corresponding to ~33.9 g/day for a 70 kg human [91].
Although long-term exposure data from the ongoing NOPARK and
NOPARK-extension trials is expected by the end 2025, it is likely wise
to avoid administering high-dose NAD-augmentation treatment in people
with active cancer disease.
Finally,
a recent epidemiological study indicated that the downstream NAD
metabolites Me-6-PY and Me-4PY may be associated with an increased risk
for major cardiovascular events in high-risk populations [92].
However, because this study was based on epidemiological association,
data from long-term, prospective interventions will be necessary to
assess whether there is a causal relationship between these metabolites
and increased cardiovascular risk. Notably, trials investigating NAD
supplementation as a treatment of heart disease have not reported such
adverse events [93].
Nevertheless, other pathways of NAD augmentation that may avoid the
generation of methylated pyridones, including inhibition or
downregulation of NADase CD38, or of purine nucleoside phosphorylase
(PNP) which affects NR degradation to Nam, are now being explored as
potential alternatives to increase NAD levels.
Concluding remarks and future perspectives
Although
the field of clinical NAD research in NDDs is advancing rapidly, it
remains in its infancy, and significant knowledge gaps and challenges
must be addressed to sustain and build on current momentum (see Outstanding questions).
Achieving sufficiently high doses in the brain is vital to properly
evaluate the therapeutic effect of NAD augmentation in neurological
diseases. It is not yet fully understood which pathways are involved in
increasing brain NAD levels, especially in humans, or which
intermediates (nucleosides, mononucleotides, or the dinucleotide itself)
penetrate the brain. Furthermore, although oral precursor
supplementation is a convenient delivery route, it would be beneficial
to establish CNS-specific NAD augmentation approaches, for example via
vehicle platforms, and some promising efforts have already been made [94].
A
recurring observation in human trials is a substantial interindividual
heterogeneity in both physiological NAD levels and the response to NAD
augmentation treatment. This includes some participants who appear to be
non-responsive in terms of cerebral NAD elevation based on 31P-MRS
data. Several factors may contribute to this heterogeneity, including
genetic predisposition and the gut microbiome composition, both of which
may influence precursor uptake and utilization. Understanding these
underlying mechanisms will be crucial for implementing personalized
treatment strategies.
A
key challenge in the field is the heavy reliance on animal models to
study the effects of NAD supplementation. Although these models can be
useful for hypothesis generation and for uncovering downstream
mechanistic insights into treatment effects, they do not accurately
reflect NDDs and have very limited, if any, translation potential to
human treatments. Therefore, current models cannot predict treatment
responses in humans. An important cautionary lesson in that regard is
offered by PD, where all (>60) trials of potential DMTs to date have
produced negative results, despite the fact that most had highly
encouraging findings in preclinical studies [95,96].
Thus, only clinical trials can provide reliable evidence of potential
disease-modifying properties of NAD augmentation therapy in NDDs. In
that regard, the currently completed trials are mostly encouraging. Most
importantly, larger and longer clinical trials are ongoing which are
designed to provide definite answers to the question that matters the
most – is NAD augmentation a successful disease-modifying strategy for
NDDs?Outstanding questions
Acknowledgments
This work is supported by grants from the Norwegian Research Council (288164), Stiftelsen Kristian Gerhard Jebsen (SKGJ-MED-023), and the Western Norway Regional Health Authorities.
Declaration of interests
C.T.
and C.D. are listed as inventors on international patent applications
relating to the use of Nam riboside as a treatment for PD and related
disorders. These patents have been filed by the Technology Transfer
Office 'Vestlandets Innovasjonsselskap AS (VIS)' on behalf of Haukeland
University Hospital, Bergen, Norway (PCT/EP2022/067412,
PCT/EP2023/060962, EP4284387).
Cited by (0)
Glossary
Disease-modifying therapy (DMT)
targets
the underlying pathology and pathophysiology of a disease to alter its
trajectory. A successful DMT should slow disease progression, arrest it
entirely, or even reverse the disease process. In the context of NDDs,
DMTs are often associated with neuroprotection with the aim of
preserving neuronal structure and function. DMTs differ fundamentally
from symptomatic treatments, which provide temporary relief of symptoms
without affecting the long-term progression or underlying mechanisms of
the disease.
Mitochondrial respiratory chain (MRC)
an
electron transport chain located in the inner mitochondrial membrane,
consisting of four protein complexes that accept and transfer electrons
from reduced NADH (at complex I) and FADH2 (at complex II) to
the final electron acceptor oxygen. Complexes I, III, and IV of the MRC
pump protons from the mitochondrial matrix into the intermembrane
space, thus converting energy that is released during electron transfer
into an electrochemical gradient over the inner mitochondrial membrane.
The gradient is degraded by ATP synthase (complex V) and the released
energy stored as newly formed ATP.
Neurodegenerative disease (NDD)
a
disorder characterized by progressive degeneration and death of neurons
in the central and/or peripheral nervous systems, resulting in
progressive loss of function, structure, and connectivity of neuronal
networks. This process typically leads to progressive neurological
impairments that may include cognitive, motor, autonomic, sensory, and
other functions, depending on the specific neural networks and brain
regions affected.
Neurodegenerative proteinopathy
NDDs
that are characterized by pathological aggregation of specific
misfolded proteins such as α-synuclein, tau, amyloid-β, and TDP43 in the
nervous system. These proteins often maintain an unstructured monomeric
form in healthy conditions but in the disease state undergo a
conformational, structural change leading to oligomerization and
subsequent aggregate formation. These aggregates can be intracellular
(e.g., Lewy bodies and neurofibrillary tangles) or extracellular (e.g.,
amyloid-β), and are generally assumed to be directly damaging to neurons
and to contribute to the pathogenesis of neurodegeneration, although
this is not fully proven.
Proteostasis
also
known as protein homeostasis, proteostasis is the maintenance of
physiological balance in the proteome, and includes protein synthesis,
folding, trafficking, and degradation.