How is your doctor going to use this to prevent your dementia and Alzheimers risk post stroke? Or is your doctor doing nothing in this problem?
Your risk of dementia, has your doctor told you of this?
1. A documented 33% dementia chance post-stroke from an Australian study? May 2012.
2. Then this study came out and seems to have a range from 17-66%. December 2013.`
3. A 20% chance in this research. July 2013.
4. Dementia Risk Doubled in Patients Following Stroke September 2018
What is your doctor's EXACT PROTOCOL TO PREVENT DEMENTIA?
The latest here:
A large-scale genome-wide cross-trait analysis reveals shared genetic architecture between Alzheimer’s disease and gastrointestinal tract disorders
Communications Biology volume 5, Article number: 691 (2022)
Abstract
Consistent with the concept of the gut-brain phenomenon, observational studies suggest a relationship between Alzheimer’s disease (AD) and gastrointestinal tract (GIT) disorders; however, their underlying mechanisms remain unclear. Here, we analyse several genome-wide association studies (GWAS) summary statistics (N = 34,652–456,327), to assess the relationship of AD with GIT disorders. Findings reveal a positive significant genetic overlap and correlation between AD and gastroesophageal reflux disease (GERD), peptic ulcer disease (PUD), gastritis-duodenitis, irritable bowel syndrome and diverticulosis, but not inflammatory bowel disease. Cross-trait meta-analysis identifies several loci (Pmeta-analysis < 5 × 10−8) shared by AD and GIT disorders (GERD and PUD) including PDE4B, BRINP3, ATG16L1, SEMA3F, HLA-DRA, SCARA3, MTSS2, PHB, and TOMM40. Colocalization and gene-based analyses reinforce these loci. Pathway-based analyses demonstrate significant enrichment of lipid metabolism, autoimmunity, lipase inhibitors, PD-1 signalling, and statin mechanisms, among others, for AD and GIT traits. Our findings provide genetic insights into the gut-brain relationship, implicating shared but non-causal genetic susceptibility of GIT disorders with AD’s risk. Genes and biological pathways identified are potential targets for further investigation in AD, GIT disorders, and their comorbidity.
Introduction
Alzheimer’s disease (AD) is the most prevalent form of dementia, characterised by neurodegeneration and a progressive decline in cognitive ability1,2. The disorder ranks as a subject of increasing global public health importance with consequences for wide-ranging social and economic adverse impacts on sufferers, their families, and the society at large1. By the year 2030, over 82 million people—and about 152 million by 2050—are projected to suffer from AD1,2. While AD has no known curative treatments, and its pathogenesis is yet to be clearly understood, a comprehensive assessment of its shared genetics with other diseases (comorbidities) can provide a deeper understanding of its underlying biological mechanisms and enhance potential therapy development efforts.
Several studies have reported a pattern of co-occurrence of dementia (and AD in particular) with certain gastrointestinal tract (GIT) disorders, microbiota, dysbiosis or medications commonly used in the treatment of peptic ulcer disease (PUD)3,4,5,6,7,8,9,10. For example, an observational study reported more than twice the odds of dementia in individuals with gastritis (adjusted odds ratio [AOR]: 2.42, P < 0.001, 95% confidence interval [CI]: 1.68–3.49)3. Another observational study found a significant association between regular use of proton-pump inhibitors (PPI, medications for gastritis duodenitis, gastroesophageal reflux disease [GERD] or PUD) and increased risk of incident dementia (hazard ratio [HR]: 1.44 [95% CI, 1.36–1.52]; P < 0.001)4. Similarly, lansoprazole (a PPI) was reported to promote amyloid-beta (Aβ) production5, the accumulation of which is central to one of the core hypotheses for the development of AD11. More recently, a longitudinal study reported more than a sixfold increased risk of AD in individuals with inflammatory bowel disease (IBD) [HR: 6.19, 95%CI: 3.31–11.57], predicting over five-fold increased incidence across all forms of dementia7.
The available evidence, thus, suggests comorbidity or some forms of association between AD and GIT disorders, although it is not clear whether GIT traits are risks for AD or vice versa. Regardless, these findings agree with the concept of the ‘gut–brain’ axis or the ‘gastric mucosa–brain’ relationship, which has been implicated between GIT-related traits and central nervous system (CNS) disorders including depression and Parkinson’s disease12,13,14,15,16,17. A relationship between AD and GIT disorders or their comorbidity can worsen the quality of life of sufferers while contributing to increased healthcare costs.
Despite the increasing number of studies reporting an association between AD and GIT traits, the biological mechanism(s) underlying this potential association remains unclear. Moreover, contrasting evidence exists7,18,19, leading to a longstanding debate on the potential links of GIT traits to the risk of AD15,18,19,20. Large-scale genome-wide association studies (GWAS), identifying an increasing number of single nucleotide polymorphism (SNPs), genes, and susceptibility loci, have been conducted separately for AD and a range of GIT traits21,22,23,24. Findings from these GWAS provide compelling evidence for the roles of genetics in the aetiologies of AD and GIT disorders including GERD, PUD, PGM (a combination of disease-diagnosis of PUD and/or GERD and/or corresponding medications and treatments—a potential proxy for PUD or GERD), gastritis-duodenitis, irritable bowel syndrome (IBS), diverticular disease, and IBD21,22,23,24. However, to the best of our knowledge, no study has leveraged the possible pleiotropy between AD and GIT disorders as a basis for discovering their shared SNPs, genes and/or susceptibility loci.
In this study, we analyse well-powered GWAS summary data to comprehensively assess the genetic relationship and potential causal association between AD and GIT disorders. We demonstrate a positive significant genetic overlap and correlation between AD and GERD, PUD, PGM, IBS, gastritis-duodenitis, and diverticular disease. Also, in a cross-trait GWAS meta-analysis, we identify many loci shared by AD and GIT disorders. Causality assessment reveals no evidence for a significant causal association between AD and GIT disorders. However, we identify shared genes reaching genome-wide significance for AD and GIT disorders in gene-based association analyses. Lastly, pathway-based analyses show significant enrichment of lipid metabolism, autoimmunity, lipase inhibitors, PD-1 signalling and statin mechanisms, among others, for AD and GIT traits.
Results
Figure 1 presents a schematic workflow for this study. Briefly, we performed three broad levels of analyses—SNP-level, gene-level, and pathway-based analyses. First, we used the linkage disequilibrium score regression (LDSC)25 to estimate the genetic correlation between AD and GIT traits, and the ‘SNP effect concordance analysis’ (SECA)26 method for concordance in SNP risk effect assessment. Second, to identify SNPs and susceptibility loci shared by AD and GIT disorders, we carried out GWAS meta-analyses. We also applied the pairwise GWAS (colocalisation) method27 to identify independent genomic loci with shared genetic influence on AD and GIT disorders. Third, using the Mendelian randomisation (MR)28 and the Latent Causal Variable (LCV)29 methods, we assessed potential (and partial) causal associations between AD and GIT disorders. Lastly, we performed gene and pathway-based analyses to identify shared genes reaching genome-wide significance and biological pathways for AD and GIT disorders. The largest publicly available AD summary statistics and GIT summary data from research consortia or public repositories were utilised for analysis (Table 1 and Supplementary Data 1).
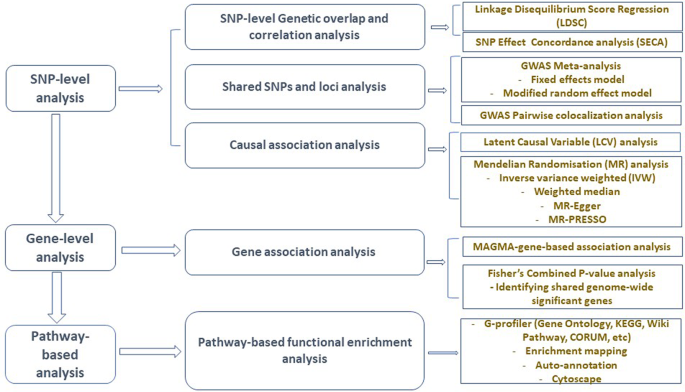
GWAS genome-wide association studies, SNP single-nucleotide polymorphism, SECA SNP effect concordance analysis, LDSC linkage disequilibrium score regression, LCV latent causal variable, MAGMA multi-marker analysis of genomic annotation, MR Mendelian randomisation, MR-PRESSO Mendelian randomisation pleiotropy residual sum and outlier, KEGG Kyoto Encyclopedia of Genes and Genomes.
Genetic correlation between AD and GIT disorders
We assessed and quantified the SNP-level genetic correlation between AD and GIT disorders using the LDSC25 analysis method. The apolipoprotein E (APOE) region has a large effect on the risk of AD; hence, we excluded APOE and the 500 kilobase (kb) flanking region (hg19, 19:44,909,039–45,912,650) from the AD GWAS. We also excluded SNPs in the 26 to 36 megabase region of chromosome six from the data given the complex LD structure in the human major histocompatibility complex (MHC). Notably, in analyses both with and without the APOE region, LDSC reveals a significant genetic correlation between AD and GIT traits (Table 2). Genetic covariance intercept estimates were not significantly different from zero (Supplementary Data 2), indicating no sample overlap between our AD and GIT GWAS.
We found a positive and significant genetic correlation (rg) of AD (excluding APOE region) with GERD (rg = 0.25, P = 8.19 × 10−18), PUD (rg = 0.28, P = 3.70 × 10−7), PGM (rg = 0.22, P = 2.38 × 10−14), gastritis-duodenitis (rg = 0.24, P = 2.40 × 10−8), IBS (rg = 0.19, P = 1.10 × 10−4), and diverticular disease (rg = 0.15, P = 2.97 × 10−5). However, we found no evidence of a significant genetic correlation between AD and IBD (rg = 0.07, P = 9.94 × 10−2) [Table 2], which may be because of the relatively small cases and sample size of the IBD GWAS (Table 1 and Supplementary Data 1). Our estimates of effective sample size (Supplementary Data 1) suggest the IBD GWAS was underpowered compared to other GIT data sets. We reproduced a pattern of a positive and significant genetic correlation between AD21 and the replication set of GIT traits with or without the APOE region, except for IBD (Supplementary Data 3).
SNP effect concordance analysis (SECA) results
Using the SECA method26, we assessed the directions of SNP-level genetic overlap between AD and GIT disorders. We provide a more comprehensive description of SECA in the methods section. Briefly, SECA performs a bi-directional analysis, assessing concordance in the direction of the effect of AD-associated SNPs (data set 1) on each of the GIT disorders (data set 2) and vice versa. First, we conducted two rounds of P-value informed LD clumping (first clumping: -clump-r2 0.1, -clump-kb 1000; second clumping: -clump-r2 0.1, -clump-kb 10000) using PLINK 1.9030. SECA subsequently assesses (using Fisher’s test) the presence of excess SNPs in which the direction of effects is concordant across 144 subsets of data set 1 (AD GWAS) and data set 2 (each of the GIT traits GWAS).
We found a positive and significant concordance of SNP risk effect across the AD (data set 1) and each of the GIT GWAS (data set 2) including IBD (Table 3). For example, of the total 144 SNP subsets tested with AD as data set 1 (Table 3), all 144 (for GERD, PGM and gastritis-duodenitis), 139 (PUD), 133 (IBS), 130 (diverticulosis) and 42 (IBD) produced Fisher’s exact tests with at least nominally significant effect concordance (odds ratio [OR] > 1 and P < 0.05). The empirical P values (Ppermuted) for the significant associations, adjusting for the 144 SNP subsets tested (using permutations of 1000 replicates), range from 0.001 to 0.018 (Table 3). These results are significantly more than expected by chance, supporting evidence of genetic overlap between AD and the GIT traits.
More at link.
No comments:
Post a Comment