Does your competent? doctor and hospital have enough functioning brain cells to see that this might help in stroke and get further research done in humans? Oh, you don't have a functioning stroke doctor or hospital, do you?
CCL5 is essential for axonogenesis and neuronal restoration after brain injury
Abstract
Background
Traumatic brain injury (TBI) causes axon tearing and synapse degradation, resulting in multiple neurological dysfunctions and exacerbation of early neurodegeneration; the repair of axonal and synaptic structures is critical for restoring neuronal function(Sounds exactly like the needs in stroke). C-C Motif Chemokine Ligand 5 (CCL5) shows many neuroprotective activities.
Method
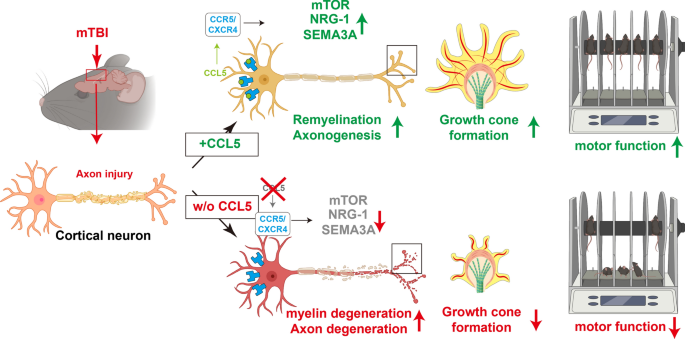
A close-head weight-drop system was used to induce mild brain trauma in C57BL/6 (wild-type, WT) and CCL5 knockout (CCL5-KO) mice. The mNSS score, rotarod, beam walking, and sticker removal tests were used to assay neurological function after mTBI in different groups of mice. The restoration of motor and sensory functions was impaired in CCL5-KO mice after one month of injury, with swelling of axons and synapses from Golgi staining and reduced synaptic proteins-synaptophysin and PSD95. Administration of recombinant CCL5 (Pre-treatment: 300 pg/g once before injury; or post-treatment: 30 pg/g every 2 days, since 3 days after injury for 1 month) through intranasal delivery into mouse brain improved the motor and sensory neurological dysfunctions in CCL5-KO TBI mice.
Results
Proteomic analysis using LC-MS/MS identified that the “Nervous system development and function”-related proteins, including axonogenesis, synaptogenesis, and myelination signaling pathways, were reduced in injured cortex of CCL5-KO mice; both pre-treatment and post-treatment with CCL5 augmented those pathways. Immunostaining and western blot analysis confirmed axonogenesis and synaptogenesis related Semaphorin, Ephrin, p70S6/mTOR signaling, and myelination-related Neuregulin/ErbB and FGF/FAK signaling pathways were up-regulated in the cortical tissue by CCL5 after brain injury. We also noticed cortex redevelopment after long-term administration of CCL5 after brain injury with increased Reelin positive Cajal-Rerzius Cells and CXCR4 expression. CCL5 enhanced the growth of cone filopodia in a primary neuron culture system; blocking CCL5’s receptor CCR5 by Maraviroc reduced the intensity of filopodia in growth cone and also CCL5 mediated mTOR and Rho signalling activation. Inhibiting mTOR and Rho signaling abolished CCL5 induced growth cone formation.
Conclusions
CCL5 plays a critical role in starting the intrinsic neuronal regeneration system following TBI, which includes growth cone formation, axonogenesis and synaptogensis, remyelination, and the subsequent proper wiring of cortical circuits. Our study underscores the potential of CCL5 as a robust therapeutic stratagem in treating axonal injury and degeneration during the chronic phase after mild brain injury.
Graphical Abstract
Introduction
Traumatic brain injury (TBI) is a complex disorder caused by external forces and is also the most significant cause of death and disability for people under the age of 40 [23, 31]. According to the Glasgow Coma Scale (GCS), TBI can be clinically divided into mild (GCS:14–15), moderate (GCS:9–13) and severe (GCS:3–8). Mild TBI is the most common brain injury caused by contact sports (i.e., hockey, football), motor vehicle accidents, and falls. Evidence from long-term studies on soldiers and athletes suggest that mild and repeated mild brain injuries are associated with the development of Chronic Traumatic Encephalopathy (CTE) [19, 37, 44], and also increase the risk of early onset Parkinsonism [18], dementia, and Alzheimer’s disease (AD) [25, 48].
Axonal shredding and tearing upon the impact, also called traumatic axonal injury (TAI) or diffuse axonal idiopathic injury (DAI), are the initial direct damages from mechanical impact on brain tissue. TAI is characterized by impaired axoplasmic transport, axonal swelling, disconnection with small hemorrhagic and/or non-hemorrhagic lesions, and brain swelling [7]. Unfortunately, much evidence suggests that neurons within the adult mammalian CNS cannot regenerate their axons after injury [13, 43]. Several deleterious cascades will be activated, leading to axon degeneration after injury. First, damaged glial cells/oligodendrocytes may not be able to provide sufficient energy to support neurons and cause neuron degeneration after injury [33, 49]. Second, increased calcium (Ca2+) influx leads to neuron apoptosis and degeneration [33, 49]. Third, glia scar and myelin-associated inhibitory proteins, such as Nogo, myelin-associated glycoprotein (Mag), and oligodendrocyte myelin glycoprotein (OMgp), can inhibit axonal regeneration after injury [15].
Recent evidence suggests that CNS neurons can revert to an embryonic-like growth state in the chronic/remodeling phase of injury to facilitate axon regeneration [46]. This “redevelopment” state provides a permissive microenvironment and the intracellular machinery for axon regrowth [9]. The endogenous repair systems activate and repair damaged axons within the chronic phase after axonal injury. The molecular machinery underlying axon regeneration is very similar to axon growth, such as Rho-GTPases Cdc42, Rac-1, and RhoA, phosphoinositide 3-kinase (PI3K)/AKT signaling pathways, and mitogen-activated protein kinase (MAPK) signaling [38]. Importantly, Rho is also the target molecule for myelin-associated inhibitory proteins [15]. PI3K/Akt activates its downstream molecules - the mammalian target of rapamycin (mTOR) and promotes axonal regeneration, synaptic plasticity, and neuronal survival after injury [35, 45]. Modulating the mTOR pathway is a novel strategy to promote axon regeneration [10]. Among three types of MAP kinases, the Erk cascade is a major pro-survival intracellular signaling pathway that can be activated by GDNF (glial cell-line derived neurotrophic factor) to promote neurite outgrowth in the spinal cord [29].
Chemokine CCL5; C-C motif ligand 5, also known as RANTES (regulated on activation, normal T cell expressed and secreted), shows many protective roles after neuronal damage, such as in stroke [51] and AD [24, 28, 32, 53]. In brain trauma, the plasma level of CCL5 increases in both human patients and animals after injury [2, 20]. Interestingly, CCL5 was found to be raised around the axonal transection site in mice immediately after injury [3]. The increased CCL5 around the axon transection site induces leukocyte infiltration to the injury site, but its function on neurons is still unclear. Our previous study identified that CCL5 is an essential factor in activating glutathione peroxidase 1 (GPX1) after brain injury, consequently reducing oxidative stress and protecting hippocampal neurons from oxidative stress-induced death; this effect facilitated memory-cognition recovery in mice after mild brain injury [22]. We also showed that CCL5 contributes to hippocampal neurons’ ATP generation and synaptic complex formation [1]. In addition, CCL5 expression after spinal cord injury is associated with axon regeneration and immune suppression [57]. The plasma level of CCL5 is highly correlated with BDNF, EGF (epidermal growth factor), and VEGF (vascular endothelial growth factor) after stroke [51]; those trophic factors contribute to neuron growth and proper brain function. Taken together, these findings suggest that CCL5 can help neurons survive from energy shortage and promote axon and synapse regrowth after injury.
In the present study, we identified that this chemokine has a robust and attractive effect on neuron growth cone formation, axon growth and myelination during post-injury repair. We specifically demonstrate for the first time that these effects are related to the ability of neuronal CCL5 to promote axon growth cone formation, axonogenesis and myelination through P70S6/mTOR signaling and the NRG1/ErbB and FGF pathways, ultimately augmenting neuronal axon and synapse regrowth after injury. Together, CCL5 promotes recovery of axogenesis and neurogenesis after brain injury.
No comments:
Post a Comment