Sounds like this is describing the neuronal cascade of death. But I see nothing here that suggests that the stroke strategy will be updated and followup occur.
The Inflammatory Response After Ischemic Stroke: Targeting β2 and β1 Integrins
 Danielle N. Edwards1,2 and
Danielle N. Edwards1,2 and  Gregory J. Bix3,4*
Gregory J. Bix3,4*- 1Sanders–Brown Center on Aging, University of Kentucky, Lexington, KY, United States
- 2Department of Neuroscience, University of Kentucky, Lexington, KY, United States
- 3Department of Neurology, University of Kentucky, Lexington, KY, United States
- 4Department of Neurosurgery, University of Kentucky, Lexington, KY, United States
Ischemic stroke is a leading cause of death and
disability with limited therapeutic options. Resulting inflammatory
mechanisms after reperfusion (removal of the thrombus) result in
cytokine activation, calcium influx, and leukocytic infiltration to the
area of ischemia. In particular, leukocytes migrate toward areas of
inflammation by use of integrins, particularly integrins β1 and β2.
Integrins have been shown to be necessary for leukocyte adhesion and
migration, and thus are of immediate interest in many inflammatory
diseases, including ischemic stroke. In this review, we identify the
main integrins involved in leukocytic migration following stroke (αLβ2, αDβ2, α4β1, and α5β1) and targeted clinical therapeutic interventions.
Introduction
Ischemic stroke is a leading cause of death and
disability in the United States with limited therapeutic interventions
available, including tissue plasminogen activator (t-PA) and
endovascular mechanical thrombectomy (Rao et al., 2014; Benjamin et al., 2017; Rai et al., 2017).
These interventions are focused on the removal of the thrombus,
restoring blood flow, oxygen and glucose to hypoperfused areas, but are
unable to affect the inflammatory, necrotic, and blood-brain barrier
(BBB) mechanism that follow. In particular, the initial inflammatory
cascade is initiated by the decrease in ATP production, release of
cytokines, influx of intracellular calcium, reactive oxygen species,
etc., that develops during occlusion and continues for days afterward (Sandoval and Witt, 2008).
Using shear forces from cerebral blood flow, marrow-derived leukocytes
(including polymorphonuclear leukocytes (PMNs), neutrophils, lymphocytes
and monocytes) are recruited to the site of injury (Dereski et al., 1993; del Zoppo, 1994; Stefanidakis and Koivunen, 2006).
For the purpose of this review, we will focus on the
recruitment and rolling of leukocytes under the direction of integrins,
as well as some of their ligands following reperfusion after ischemic
stroke. We will then introduce recent β2 and β1 integrin-specific stroke clinical trials, and, finally, discuss potential future directions for the field.
Role of Integrins Post-Stroke: an Overview
Integrins are a diverse group of heterodimers composed of
18 different α and β subunits, creating 24 unique combinations.
Integrins exist on every cell type, while exhibiting a high diversity of
ligands and grouped into four different receptor groups: RGD
(Arg-Gly-Asp), laminin receptors, collagen receptors, and
leukocyte-specific receptors. Within these groups, integrins can have a
variety of ligands and roles following ischemic stroke (reviewed in Edwards and Bix, 2019). Under normal cerebrovascular conditions, integrins are in a highly inactive state, typically in a bent conformation (Takagi et al., 2002; Nishida et al., 2006).
Following ischemic stroke, activation signals are sent. Chemokines are
translocated to the lumen, on the apical side of endothelial cells, to
induce “inside-out” signaling (Chavakis, 2012).
Integrins then undergo a conformational change to increase integrin
affinity for potential ligands while enhancing detection by localizing
to the leading or rear-facing edge of the leukocyte’s cell wall for
ligand detection (Ridley et al., 2003; Hyun et al., 2009).
Activated integrins then bind to available ligands, permitting
leukocytic rolling and intracellular signaling. This is termed
“outside-in” signaling (Hato et al., 1998; Tominaga et al., 1998; Ley et al., 2007).
Leukocytes continue movement to the site of injury, looking for areas
to cross the endothelial cell barrier, and eventually coming to a halt.
Aggregation/clustering of integrins increases binding avidity (strength
of binding), preventing flow conditions from detaching leukocytes from
the endothelial cells (Ley et al., 2007). Using transmigration, leukocytes will infiltrate into the cerebral parenchyma using these integrin-ligand connections.
β2 Integrins
β2 integrins are the only group of integrins exclusively expressed on leukocytes (derived from hematopoietic cells) (Schenkel et al., 2004), and like most integrins, are highly conserved across species (Schittenhelm et al., 2017).
They are also the most highly expressed integrin on circulating blood
leukocytes, tending to cluster at the retraction area of the cell (the
rear), in both an active and inactive state, compared to other β1, β4, β3, and β7 integrins found on circulating leukocytes (Pierini et al., 2000; Lindbom and Werr, 2002). Genetic leukocyte adhesion changes (LAD-1, as discussed above) has been attributed to mutations in the β2 subunit, reducing β2 expression. Thus, leukocytic movement is reduced on the cell surface with less movement toward the site of inflammation (Arnaout, 1990; Scharffetter-Kochanek et al., 1998). Importantly, in β2 inhibited mice, there is not total arrest of leukocytic recruitment or infiltration (Pierini et al., 2000), suggesting that other factors likely play a role. There are 4 identified heterodimers of β2 integrins, and of these, the most highly studied are αLβ2 and αMβ2 in ischemic stroke, and will be reviewed in more detail below. The other β2 integrins, αXβ2 and αDβ2, have not been individually studied in the context of stroke as have αLβ2 and αMβ2 integrins, though CD18 (β2)
inhibition in addition to t-PA has been shown to increase the time
window of t-PA administration without an increase in hemorrhagic
transformation in a rat embolic stroke model (Zhang et al., 1999). Furthermore, Figure 2 summarizes the results in this section.
FIGURE 2
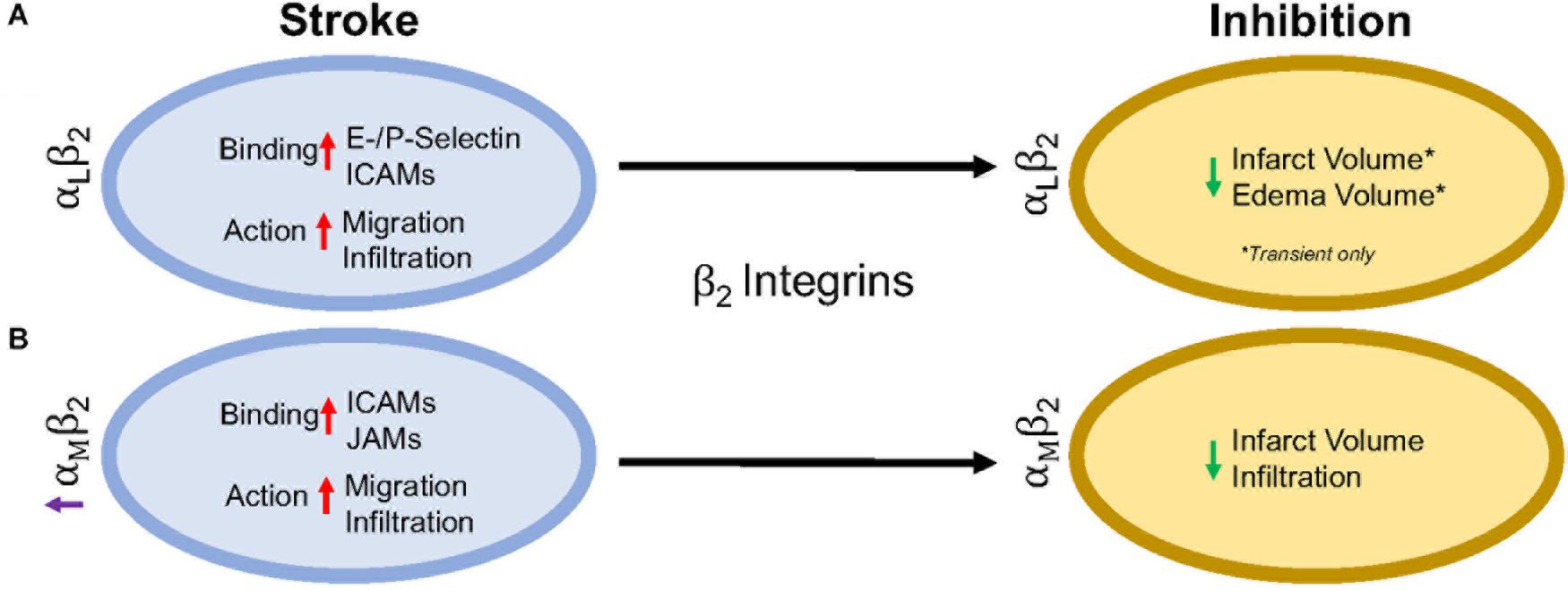 Figure 2. Representative image of the β2 integrin response following experimental stroke and inhibitory antibody treatment in preclinical trials. Inhibition of (A) αLβ2 and (B) αMβ2 integrins post-stroke responses and effects.
Figure 2. Representative image of the β2 integrin response following experimental stroke and inhibitory antibody treatment in preclinical trials. Inhibition of (A) αLβ2 and (B) αMβ2 integrins post-stroke responses and effects.αLβ2 Integrin
Integrin αLβ2 is also referred to as CD11a/CD18 and LFA-1 (lymphocyte functional-associating antigen-1). αLβ2
integrin acutely increases in ischemic stroke patients, with detectable
amounts through 72 h associated around the area of ischemia (Gerhard et al., 2000; Zhao et al., 2002). This suggests a correlation between αLβ2 integrin expression and inflammatory damage following ischemia. αLβ2 is expressed on all leukocytes (Soriano et al., 1999), though at particularly high levels on T-lymphocytes (Hammond et al., 2014; Walling and Kim, 2018). In healthy individuals, extracted blood analysis revealed that αLβ2 activation requires leukocytic rolling on P- or E-selectins, inducing an active conformational change (Kuwano et al., 2010),
but it is the binding of chemokines g-protein coupled receptors (GPCR)
and Rap-1 activation that induces the high-affinity conformational state
of αLβ2 (Steffen et al., 1994; Greenwood et al., 1995; Ghandour et al., 2007). In this state, αLβ2 has many possible ligands, ICAM-1, ICAM-2, ICAM-3, ICAM-4, ICAM-5, and junctional adhesion molecule-1 (JAM-1) (Marlin and Springer, 1987; de Fougerolles et al., 1991, 1994; Tian et al., 2000), though ICAM-1 is preferentially bound (Walling and Kim, 2018). The high avidity αLβ2-ICAM-1
complex, once formed, allows t-lymphocytes to move against circulatory
flow and the shear forces, resulting in the high-speed movement of
leukocytes (Katakai et al., 2013; Dominguez et al., 2015).
In an intraluminal model of experimental ischemic stroke, αLβ2
inhibition with the use of transgenic mice results in reduced infarct
volume, edema volume and mortality. However, this phenomenon is evident
in transient, but not permanent middle cerebral artery occlusion (Arumugam et al., 2004). This may be due to the previously mentioned high avidity of αLβ2-ICAM-1 bonds, and is evident in an in vitro study using αLβ2
(LFA-1) knock-in mice that experience high avidity through binding of
lymphocytes mediated through ICAM-1 binding, but are unable to continue
movement due to a non-polarized uropod (Park et al., 2010). An explanation for this phenomenon may be that the recycling process within the leukocyte is overwhelmed (Shaw et al., 2004). By enhancing αLβ2 expression, recycling may not be able to allow for dislocation of αLβ2-ICAM-1 complexes, preventing movement from the loss of high adhesion bonds. Enhanced αLβ2 expression could be a potential new avenue for therapy, especially if no enhanced mortality, infection, etc., are observed.
Independently, ICAMs play a significant role in
inflammation following ischemic stroke. ICAM-1, in particular, is
acutely increased in both cultured human endothelial cells undergoing
hypoxia and following intraluminal suture middle cerebral artery
occlusion, while expression remains sustained for up to a week
post-injury (Hess et al., 1994a,b; Zhang et al., 1995). ICAM-2, another possible ligand, does not change in expression following cytokine stimulation (de Fougerolles et al., 1991; Nortamo et al., 1991a,b). Furthermore, serum of ischemic stroke patients contains soluble ICAM-1, but not ICAM-2 in addition to being a risk factor (Kaplanski et al., 1994; Shyu et al., 1997).
Antibodies targeting ICAM-1 in rodents and humans have shown
contradictory results. An intraluminal suture middle cerebral artery
occlusion model in mice and rats showed a decrease in leukocyte
infiltration and infarct volume (Connolly et al., 1996; Kitagawa et al., 1998; Vemuganti et al., 2004), while one study reported opposing effects (Furuya et al., 2001).
ICAM-1 inhibition was translated to the clinic through testing of the
murine ICAM-1 antibody, Enlimomab in ischemic stroke. Unfortunately, the
study was halted early due to increased rate of infection, infarct
volumes, neurological scores and mortality for patients (Furuya et al., 2001).
αMβ2 Integrin
Integrin αMβ2, also known as CD11b/CD18 and Mac-1 (macrophage-1 antigen), exhibits many similarities to αLβ2 through its expression on all leukocytes (Springer et al., 1979), and common ligand binding partners such as the family of ICAMs and JAMs (von Andrian et al., 1991). Additional ligands are fibrinogen, heparin (von Andrian et al., 1991), elastase (Cai and Wright, 1996), complement C3 fragment (C3bi) (Micklem and Sim, 1985), kinogen components, and urokinase and its receptor (Chavakis et al., 1999). Just as αLβ2, hypoxia induced factors (cytokines, chemokines, etc.) induce conformational change of αMβ2 to a high affinity ligand-binding state (Stanimirovic et al., 1997). Binding assays with ICAM-1 as a ligand and both αLβ2 and αMβ2 as receptors show αLβ2 integrin is preferably bound (Lub et al., 1996). This suggests that the binding sites on both αLβ2 and αMβ2 compete for ICAM-1 binding.
Following experimental ischemic stroke in rats, integrin αMβ2 is upregulated (Campanella et al., 2002),
and has shown benefit when inhibited. Antibodies against both
CD11b/CD18 reduce infarct volume and reestablish cerebral blood flow as a
result of decreased neutrophil infiltration following intraluminal
stroke surgery (Chen et al., 1994; Bowes et al., 1995; Zhang et al., 1995).
In a different approach, the addition of recombinant neutrophil
inhibitory factor (rNIF) inhibits a binding domain on Mac-1 and yields
similar results in the same intraluminal occlusion model (Jiang et al., 1998). Furthermore, and similarly to αLβ2 integrin inhibition, inhibition of αMβ2 is also effective in transient, but not permanent experimental ischemic stroke in an embolic occlusion model (Zhang et al., 2003).
β1 Integrins
β1 integrins are a diverse set of integrins, with laminin-binding, collagen-binding, RGD-binding and leukocyte heterodimers. β1 integrins are not as highly expressed on leukocytes as β2 integrins, but they do play a major role in leukocyte adhesion and migration following ischemic stroke. The activity of β1 integrins is similar to β2 integrins. They undergo a conformational change to induce “inside-out” and “outside-in” cellular signaling (Campanero et al., 1994). As the cells migrate, the β1
integrins are most commonly clustered around the uropod, but will be
located in any area of the leukocyte that is in contact with the
endothelial cell or extracellular matrix (Campanero et al., 1994; Caimi et al., 2001). Inhibition of the β1 integrin, just as with β2 integrin inhibition, does not fully stop leukocyte rolling. However, when both β1 and β2 integrins are inhibited, complete leukocyte arrest occurs (Lobb and Hemler, 1994; Pierini et al., 2000). This suggests that both β1 and β2 integrins are necessary for leukocyte migration, regardless of expression load. Of all the β1 integrins, both α4β1 and α5β1 appear to be the most highly expressed and the most studied in post-stroke inflammation. The other β1 integrin expressed on leukocytes, α9β1, has not been studied in the context of stroke as its expression and role has not yet been fully elucidated in the brain. Figure 3 summarizes the results discussed in this section.
FIGURE 3
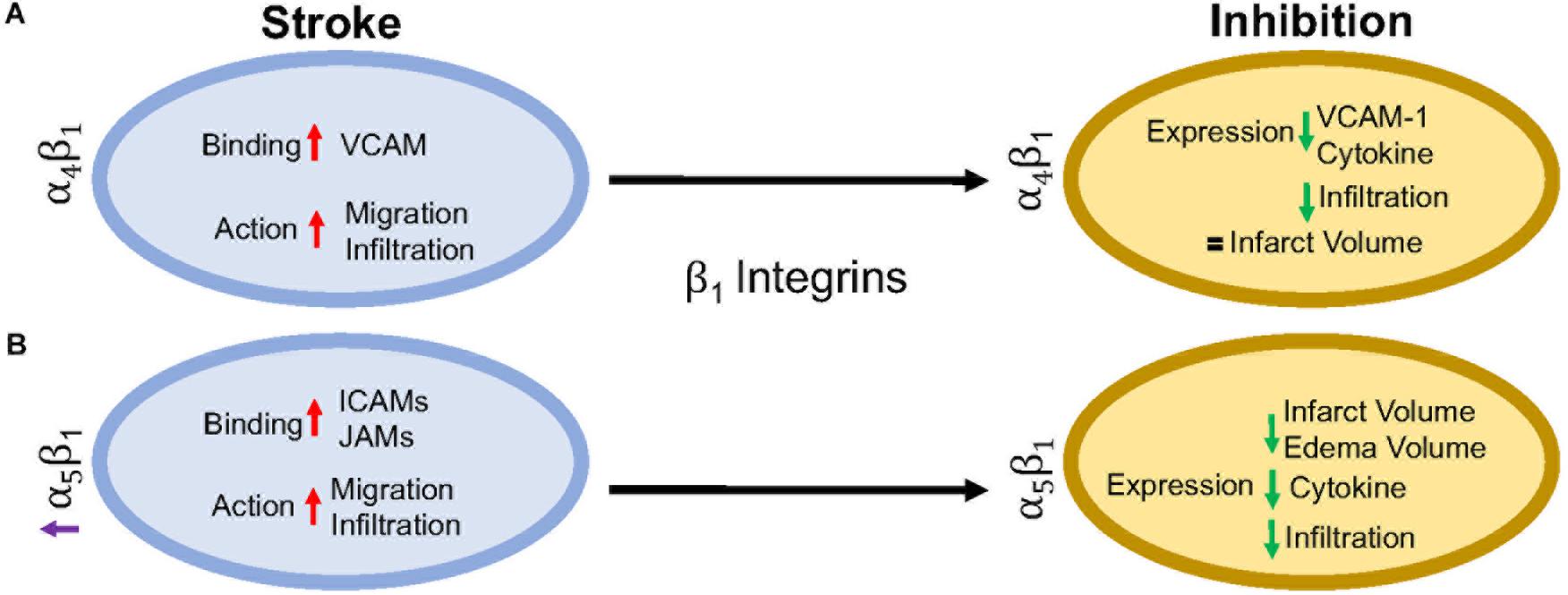 Figure 3. Representative image of the β1 integrin response following experimental stroke and inhibitory antibody treatment in preclinical trials. Inhibition of (A) α4β1 and (B) α5β1 integrins post-stroke responses and effects.
Figure 3. Representative image of the β1 integrin response following experimental stroke and inhibitory antibody treatment in preclinical trials. Inhibition of (A) α4β1 and (B) α5β1 integrins post-stroke responses and effects.α4β1 Integrin
α4β1, also known as CD49d/CD29 VLA-4 (very late antigen-4),
is localized primarily to leukocytes (neutrophils, monocytes,
lymphocytes, macrophages, etc.) and microglia as a leukocyte-specific
receptor. Additionally, α4 will also dimerize with β4, which is found in gut endothelium (Hammond et al., 2014). Activation of α4β1 integrin results from the binding of upregulated chemokines to GPCRs in the same manner as αLβ2 as discussed above (Vajkoczy et al., 2001). This stimulates binding to α4β1’s preferred ligand, VCAM-1, but experiments have shown some preference for paxillin ICAM-1 (Steffen et al., 1994; Ghandour et al., 2007), and fibronectin (Hart and Greaves, 2010) as well. Interestingly, instead of using the β1 submit of the heterodimer for binding, integrin α4β1 uses its α subunit of α4β1 to mediate binding to VCAM-1 (Luo et al., 2007).
Preclinical ischemic stroke studies targeting α4β1
have shown increasingly varied results. Most researchers reported a
decrease in VCAM-1 expression, cytokine production, and infiltrating
leukocytes (Liesz et al., 2011; Langhauser et al., 2014; Llovera et al., 2015),
but this reduction in inflammation did not result in reduced infarct
volumes or functional deficit following analysis of a randomized
preclinical trial involving six different centers (Llovera et al., 2015).
Langhauser et al went one step further and found that no treatment
paradigm (prophylactic or therapeutic) and no model (transient or
permanent) showed efficacy (Langhauser et al., 2014). On the other hand, both Becker, 2002 and Relton et al., 2000 found that inhibition of α4
improved both infarct volumes and functional deficits. When a
preclinical randomized control trial was implemented at multiple
centers, researchers found efficacy only in patients with small infarct
volumes (Llovera et al., 2015). Collectively, these contradictory results may be caused by a couple of scenarios, 1) the varying expression of integrin α4β1 expression following ischemic stroke resulting in continued leukocyte infiltration, or 2) integrin α4β1
is not a primary driver of post-stroke pathophysiology, but other
factors, including other integrins, promote leukocyte migration (Hammond et al., 2014).
α5β1 Integrin
α5β1, also known as CD49e/CD29 and
VLA-5, plays an as yet largely undetermined role in inflammation, with
studies primarily limited to cell culture. What is known is that
leukocytes express different β1 integrins with α5β1 composing around 50% of all β1 –integrins expressed on neutrophils (Pierini et al., 2000) and monocytes (Pacifici et al., 1994). Additionally, α5β1 is necessary for leukocyte adhesion. Only inhibition of both α5β1 and β2 integrins completely blocks adhesion in vitro (Pierini et al., 2000), while inhibition of α5β1 alone prevents transmigration across the BBB (Labus et al., 2018) in vitro.
As an RGD receptor, fibronectin has been shown to be the primary and
preferred [over other potential ligands such as fibrinogen (Suehiro et al., 1997)] ligand for α5β1 on endothelial cells and leukocytes (Schaffner et al., 2013; Bharadwaj et al., 2017). Importantly, in the presence of activated αLβ2, leukocyte α5β1 binding to fibronectin is enhanced (Bohnsack, 1992; Loike et al., 1999; Gronholm et al., 2016). α5β1 integrin expression is induced by cytokines, particularly TNFα (Li et al., 2011) toward the leading edge of the cell in contrast with other integrins at the uropod (Pierini et al., 2000). Furthermore, α5β1 integrin appears to be highly sensitive to calcium (Pierini et al., 2000), an ion that is increased rapidly following reperfusion (Sandoval and Witt, 2008). Upon calcium buffering, α5β1
expression moves from the front of the cell to the uropod and the
leukocyte becomes elongated. The change in expression localization and
morphology is attributed to non-movement as the leukocyte cannot detach α5β1 from the vascular wall (Pierini et al., 2000). Recently, Edwards et al. (2019) found that inhibition of α5β1
integrin by the small peptide ATN-161 prevented CD45+ leukocytes from
infiltrating the brain parenchyma following the tandem/transient common
carotid artery/middle cerebral artery occlusion model. Additionally,
mice were observed to have reduced BBB permeability, functional
deficits, edema, and infarct volume following middle cerebral artery
occlusion (Roberts et al., 2015; Edwards et al., 2019). Thus, targeting α5β1 after ischemic stroke could be a new avenue for reduction of inflammation following ischemic stroke.
Clinical Implications
The preclinical studies discussed here point towards the potential of targeting β2 and β1
integrins in the treatment of post-stroke inflammation. Though their
potential has not fully been elucidated, many clinical trials, not just
limited to stroke, have been approved in the last 10 years targeting
these integrins.
The most common target for post-stroke inflammation are the β2 integrins. Though some efficacy has been reported, no clinical stroke trials to date have targeted the αL
subunit in stroke patients. However, one clinical trial with the
monoclonal antibody, Efalizumab, has shown promise in decreasing
T-lymphocyte rolling in patients with moderate-severe plaque psoriasis (Lebwhohl et al., 2003).
In preclinical studies targeting αMβ2, a hookworm isolated recombinant glycoprotein targeting rNIF (UK279276) (Zhang et al., 2003) and humanized Hu23F2G (Leukarrest) (Yenari et al., 1998),
were both shown to decrease infarct volume and increase functional
recovery following reperfusion. Both therapies had negligible side
effects in Phase 1 studies and thus were continued to a Phase II study,
respectively, before the trials were halted due to no observed efficacy (Becker, 2002; Krams et al., 2003). The failure to target αMβ2 integrin may be due to the observation that human ischemic stroke patients do not experience the increase in αMβ2 expression as seen in rodent stroke models (Caimi et al., 2001). Interestingly, when given in conjunction with United Kingdom279276, patients experienced a slight improvement (Krams et al., 2003), but no follow-up has been conducted. This interesting effect may be worth additional investigation in future clinical trials.
Clinical inhibition of β1 integrins, on the other hand, is small and varied. Of the current clinical trials, one trial has emerged targeting α4β1 in the context of ischemic stroke. The monoclonal antibody targeting the α4 subunit (Natalizumab) has been successful in protecting patients from relapses in multiple sclerosis (Polman et al., 2006) and Crohn’s disease (Sandborn et al., 2015).
However, in a Phase II ischemic stroke study, patients receiving
Natalizumab showed no improvement in infarct growth or neurological
scores over 30 days. Furthermore, two patients (out of 79) died from
serious infections attributed to Natalizumab treatment (Elkins et al., 2017). At this time, there are no further clinical trials planned.
Future Considerations
As discussed in this review, targeting leukocytic
integrins has had limited to no efficacy in clinical trials.
Importantly, these studies have collectively employed only three
different therapeutics and two targets; there are still significant
areas that can be investigated. Though not discussed here, most
preclinical investigations have focused on the ligands themselves rather
than the integrin as the therapeutic target, highlighting the continued
importance of integrins in stroke.
It is also important to note that preclinical studies
carried out in rodents inadequately model the post-stroke
pathophysiology that patients experience. Preclinical stroke research is
also typically limited, focusing on one species, sex, and age that do
not necessarily match the demographic of stroke patients (see Kahle and Bix, 2012
for a review of this topic). Furthermore, as the changes following
stroke and/or reperfusion are inadequately understood, identifying
appropriate therapeutic targets that translate from the lab to clinical
trials, has been particularly challenging.
However, this does not suggest abandoning therapeutic
trials for ischemic stroke. As mentioned above, stroke is a leading
cause of death and disability, separate from cardiovascular disease.
This will not improve without intervention with our aging and obese
population. Fortunately, with the advent of stroke mortality-altering
therapies, i.e., t-PA and endovascular mechanical thrombectomy, our
financial burden has shifted to aftercare. When we review the amount of
trials performed for thrombolytic agents (Multicentre Acute Stroke Trial–Italy (MAST-I) Group, 1995; National Institute of Neurological Disorders and Stroke rt-Pa Stroke Study Group, 1995; The Multicenter Acute Stroke Trial–Europe Study Group, 1996) and endovascular thrombectomy [MR CLEAN (Berkhemer et al., 2015), ESCAPE (Goyal et al., 2015), EXTEND IA (Campbell et al., 2015), SWIFT PRIME (Saver et al., 2015), and REVASCAT (Jovin et al., 2015)]
as potential treatments of ischemic stroke, it is obvious that the
complexities of stroke affect the outcome of the clinical trial. This
includes, but is not limited to, the time a patient takes to arrive at
an ER, time to treatment, location of the stroke, amount of surrounding
collaterals, current medications and co-morbidities (diabetes, cancer,
etc.), and if the patient has experienced multiple strokes.
Based on current advances, as well as previous failures, a
focus on integrins as a therapeutic target for stroke is emerging. A
significant reason for this focus may be the complex, multi-dimensional
role that integrins appear to play in brain pathophysiology. Integrins
are diverse, existing on all cell types with varying roles depending
upon expression and activation. This complexity can represent a
significant challenge to integrin-targeted therapies inasmuch as such
therapies could have diverse, even unintended off-target effects.
However, we believe that this can be overcome by a better understanding
of how integrin function and expression is altered after stroke, with
the potential to exploit stroke-dependent integrin changes to
therapeutic effect. For example, identifying a specific integrin to be
upregulated in select cells in the post-stroke brain or brain-targeting
cells, but not in other organs, could render it a viable therapeutic
target. This emphasizes the need and importance of preclinical stroke
research to discover and unravel the complexities of integrin biology.
We are confident that such studies will result in viable new stroke
therapies.
Conclusion
In this review, we have implicated integrins as an area
of research for limiting inflammation following ischemic stroke. To
date, therapeutic inhibition of αLβ2, αMβ2, and α4β1
has shown promising results in preclinical studies, but translation to
the clinic has been disappointing. Going forward, more targeted
antibodies to all reactive β1 and β2 integrins
after ischemic stroke may prove more beneficial, but more research needs
to be done to completely understand the human inflammatory response and
how that relates to changes in preclinical models.
No comments:
Post a Comment