Since excitotoxicity has been known for decades our fucking failures of our stroke associations have excelled in DOING NOTHING to solve it. Yes it is hard to solve but leaders solve problems. WHERE THE FUCK ARE OUR STROKE LEADERS? Cowering somewhere they don't have to talk to survivors? You can email me, I have my address in the right column under 'email me'. See how easy it is to talk to a survivor? Or are you afraid of me?
A Neurotoxic Ménage-à-trois: Glutamate, Calcium, and Zinc in the Excitotoxic Cascade
 Alberto Granzotto
Alberto Granzotto Lorella M. T. Canzoniero
Lorella M. T. Canzoniero Stefano L. Sensi
Stefano L. Sensi- 1Sue and Bill Gross Stem Cell Research Center, University of California, Irvine, Irvine, CA, United States
- 2Center for Advanced Sciences and Technology (CAST), University “G. d’Annunzio” of Chieti−Pescara, Chieti, Italy
- 3Department of Neuroscience, Imaging, and Clinical Sciences (DNISC), Laboratory of Molecular Neurology, University “G. d’Annunzio” of Chieti−Pescara, Chieti, Italy
- 4Department of Science and Technology, University of Sannio, Benevento, Italy
- 5Institute for Memory Impairments and Neurological Disorders, University of California, Irvine, Irvine, CA, United States
Fifty years ago, the seminal work by John Olney provided the first evidence of the neurotoxic properties of the excitatory neurotransmitter glutamate. A process hereafter termed excitotoxicity. Since then, glutamate-driven neuronal death has been linked to several acute and chronic neurological conditions, like stroke, traumatic brain injury, Alzheimer’s, Parkinson’s, and Huntington’s diseases, and Amyotrophic Lateral Sclerosis. Mechanisms linked to the overactivation of glutamatergic receptors involve an aberrant cation influx, which produces the failure of the ionic neuronal milieu. In this context, zinc, the second most abundant metal ion in the brain, is a key but still somehow underappreciated player of the excitotoxic cascade. Zinc is an essential element for neuronal functioning, but when dysregulated acts as a potent neurotoxin. In this review, we discuss the ionic changes and downstream effects involved in the glutamate-driven neuronal loss, with a focus on the role exerted by zinc. Finally, we summarize our work on the fascinating distinct properties of NADPH-diaphorase neurons. This neuronal subpopulation is spared from excitotoxic insults and represents a powerful tool to understand mechanisms of resilience against excitotoxic processes.
Introduction
Excitotoxicity is a form of neuronal death triggered by excessive and/or sustained exposure to the amino acid glutamate, the primary excitatory neurotransmitter in the brain. Evidence accumulated in the past four decades indicates that excitotoxicity is a critical contributor to the neuronal demise occurring upon acute and chronic neurological conditions, like stroke, Alzheimer’s disease (AD), Huntington’s disease (HD), Amyotrophic Lateral Sclerosis (ALS), and Parkinson’s disease (PD) (Mehta et al., 2013).
Although, 50 years have passed since the first description of glutamate’s neurotoxic activity (Olney, 1969), therapeutic strategies set at counteracting these processes have been only partially exploited. In that regard, the targeting of upstream mechanisms of glutamate-driven neurotoxicity has produced, in the late 80s, an early wave of enthusiasm and fueled a level of optimism that has not been corroborated in the following years. These approaches have been found promising in preclinical models (Lee et al., 1999) but failed in clinical trials (Lee et al., 1999; Ikonomidou and Turski, 2002; Chamorro et al., 2016; Choi, 2020). Only riluzole and memantine, two drugs that target glutamate-driven neuronal death, have been approved for the treatment of ALS and AD, respectively.
Although, most of the preclinical findings failed “the bench to bed” translation, this experimental evidence has significantly helped dissect the molecular underpinnings of excitotoxicity. These studies have also helped provide support for the excitotoxic cascade hypothesis (Zivin and Choi, 1991; Choi, 2020). The construct posits that excitotoxic neuronal death is primarily mediated by the glutamate-driven activation of N-methyl-D-aspartate receptors (NMDARs) and the subsequent toxic intraneuronal accumulation of calcium (Ca2+). The NMDAR-driven Ca2+ overload is, in fact, a mandatory step in the process as most of the downstream mechanisms of the cascade, like the generation of reactive oxygen species (ROS; of mitochondrial and non-mitochondrial origin), or reactive nitrogen species (RNS), the concurrent mitochondrial dysfunction, metabolic impairment, as well as the activation of necrotic/apoptotic pathways, are all Ca2+-dependent processes (Lee et al., 1999; Lai et al., 2014; Bano and Ankarcrona, 2018; Choi, 2020; Swanson and Wang, 2020). However, Ca2+ is not alone, and other cations find a way to participate in the death banquet. Zinc (Zn2+) is, for instance, a VIP guest.
In the review, we provide a brief overview of the role of Zn2+ in the brain and discuss its neurotoxic properties and how they intertwine with the excitotoxic cascade. Finally, we focus on the distinct features of the NADPH-diaphorase neurons, a subpopulation spared from excitotoxic insults offering an intriguing model to further our understanding of neuroprotective mechanisms.
Zinc Homeostasis and Its Role in Brain Functions
After iron, Zn2+ is the most abundant metal ion in the brain. The cation can be found in either structural or labile forms (Sensi et al., 2009). Structural Zn2+ is tightly bound to proteins/peptides and acts as a critical component for proper protein folding or as the catalytic/co-catalytic element required for several enzymes (McCall et al., 2000).
Labile, free Zn2+ is either stored in the lumen of intracellular organelles, like synaptic “zinkergic” vesicles, mitochondria, lysosomes, the endoplasmic reticulum (ER), and the Golgi apparatus, or bound to metallothioneins (MTs), a class of metal-binding redox-sensitive proteins (Maret, 1994). Under physiological conditions, cytosolic Zn2+ concentrations are kept in a picomolar to a low nanomolar range (Outten and O’Halloran, 2001) through the carefully orchestrated activity of Zn2+ transporters (ZnTs), Zrt-, Irt-related proteins (ZIPs), Zn2+-stores and binding proteins (Sekler et al., 2007; Sensi et al., 2009).
Zn2+ within synaptic vesicles is released, along with glutamate, during excitatory neurotransmission (Sensi et al., 2009). Once released in the synaptic cleft, the cation shapes the post-synaptic glutamate responses by modulating the activity of glutamatergic receptors, like NMDARs and the α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors (AMPARs) (Paoletti et al., 1997; Kalappa et al., 2015). Zn2+ exerts an inhibitory effect on NMDARs by acting on a high (nanomolar) and a low (micromolar) affinity site that is located on the GluN2A and GluN2B subunits, respectively (Rachline et al., 2005). As for AMPARs, the cation has been proposed to exert an inhibitory activity by acting on the histidine residues of the receptor ligand-binding domain (Kalappa et al., 2015). However, direct structural evidence for this interaction is still missing. Of note, recent findings indicate that ZnT1-dependent cation extrusion in the proximity of synaptic NMDARs is required for the Zn2+-dependent inhibition of the receptor (Mellone et al., 2015; Krall et al., 2020). A novel post-synaptic mechanism that may put under a new light the cation’s modulating activities as the metal has been so far thought to only act via its presynaptic release. Extracellular Zn2+ is also implicated in the modulation of neurotrophic signaling as the cation is critical for the activity of the matrix metalloproteinases (MMPs), a class of enzymes involved in matrix remodeling and the maturation of the brain-derived neurotrophic factor (BDNF) from its precursor form proBDNF (Hwang et al., 2005), a process activated by Zn2+ supplementation and impaired by metal chelation (Corona et al., 2010; Frazzini et al., 2018). The proBDNF/BDNF balance is critical for neuronal functioning as the two peptides exert opposite effects. BDNF affects long-term memory processes and neuronal survival. On the contrary, proBDNF inhibits GABAergic neurotransmission (Riffault et al., 2014), facilitates long-term depression (Woo et al., 2005), and activates neuronal death-related pathways (Teng et al., 2005; Mizui et al., 2016). Zn2+ has also been shown to activate the BDNF receptor TrkB directly. This process, called transactivation, is triggered by synaptically-released Zn2+ and/or ROS-driven intracellular Zn2+ elevations (Huang et al., 2008; Huang and McNamara, 2012). However, the mechanisms described “in vitro” settings do not entirely translate in “in vivo” conditions as, surprisingly, TrkB activation has been found to be unaffected in transgenic mice devoid of vesicular Zn2+ (Helgager et al., 2014).
Unlike what is known about vesicular Zn2+, the intracellular labile pools’ physiological significance has been only partially unraveled. Along with its role as a metal reservoir, compelling evidence indicates that releasable Zn2+ can affect mitochondria and lysosomal functioning and, in close analogy with Ca2+, act as a signaling molecule (Yamasaki et al., 2007).
Most importantly, like Ca2+, when dysregulated, Zn2+ can turn into a potent neurotoxin (Sensi et al., 2009).
Zinc as Neurotoxin
The contribution of Zn2+ in neurodegenerative processes has been extensively investigated. In conditions characterized by the overactivation of excitatory signaling, synaptically released Zn2+ can flux into post-synaptic neurons through Zn2+ permeable channels (Sensi et al., 2009). Zn2+ entry occurs primarily through voltage-gated Ca2+ channels (VGCCs) and Ca2+ permeable AMPARs (CP-AMPARs) (Sensi et al., 1997, 1999b; McDonald et al., 1998; Colvin et al., 2000; Sheline et al., 2002). AMPAR permeability to Ca2+ and Zn2+ ions is restricted to certain neuronal populations or occurs upon disease associated challenges. The distinct expression pattern and the high permeability to Ca2+ and Zn2+ contribute to the unique role of CP-AMPAR in selective neurodegeneration (see Weiss, 2011 for an extensive review on the topic). NMDARs are poorly permeable to Zn2+ ions (Sensi et al., 1997).
Additional routes of entry are the Na+-Zn2+ exchanger and transient receptor potential channels (TRP); however, their contribution to the cation’s toxic accumulation is still mostly unexplored (Bouron and Oberwinkler, 2014). The exact amount of Zn2+ released from presynaptic terminals and the degree of its contribution to cation accumulation in the post-synaptic neurons are also not completely clear and have been matters of lively debates (Kay and Toth, 2008; Vergnano et al., 2014).
Zn2+ released from intracellular pools also participates in the cation’s cytosolic build-up (McCord and Aizenman, 2014). In this regard, MTs are a significant source of intracellular Zn2+ (Maret, 1994; Aizenman et al., 2000). MTs mobilize a large amount of Zn2+ (ranging 10–100 nM) in response to Ca2+-driven generation of ROS/RNS as well as in conditions of mild acidosis, a combination found in excitotoxic settings and several neurodegenerative conditions. The critical role played by Zn2+ released from MTs in the production of neuronal death is supported by the ability of oxidative agents [i.e., 2,2′-dithiodipyridine (DTDP) or N-ethylmaleimide (NEM)] to trigger widespread apoptotic neuronal death (Aizenman et al., 2000; Gibon et al., 2010). This process is mostly reduced by Zn2+chelators and independent of Ca2+ load. Intraneuronal Zn2+ rises are not the final step of the toxic cascade but are critical to trigger mitochondrial and lysosomal dysfunction, as well as the activation of neurotoxic pathways in the cytosol (Sensi et al., 2009; Ji et al., 2019; Koh et al., 2019).
Mitochondria are a primary target of intracellular Zn2+ as the cation accumulates in the organelles thanks to their steep electrochemical gradient (Δp). Once sequestered, Zn2+, along with Ca2+, contributes to Δp loss and promotes ROS generation (Sensi et al., 1999a; Ji and Weiss, 2018). Zn2+ mobilization is an essential prerequisite to trigger irreversible mitochondrial dysfunction as the cation, by acting in close synergy with Ca2+ damaging effects, promote the full demise of the organelles and, eventually, cell death (Jiang et al., 2001; Granzotto and Sensi, 2015). Within mitochondria, Zn2+ acts by inhibiting complexes of the electron transport chain (ETC) and α-ketoglutarate dehydrogenase (αKGDH) of the Krebs cycle, thereby promoting aberrant ROS production and metabolic failure (Sensi et al., 2009; Ji et al., 2019). Zn2+ interactions with αKGDH and the matrix-facing complexes of the ETC support the presence of the cation in the mitochondrial matrix. Moreover, recent findings indicate that mitochondrial Zn2+ uptake through the activation of the mitochondrial Ca2+ uniporter (MCU) participates in producing the neuronal death found in preclinical models of brain ischemia (Ji et al., 2019, 2020). Zn2+ also triggers the permeabilization of the mitochondrial membrane through the activation of the mitochondrial permeability transition pore (MPTP; a key promoter of cell death; Bernardi et al., 2015), thereby generating the release/production of pro-apoptotic factors [like cytochrome c, apoptosis-inducing factor (AIF), and ΔN-Bcl-XL] (Jiang et al., 2001; Bossy-Wetzel et al., 2004; Bonanni et al., 2006; Ji et al., 2019).
In addition, Zn2+ elevations target lysosomes (Koh et al., 2019). Lysosomal Zn2+ rises, coupled with the accumulation of lipid peroxidation byproducts (4-hydroxinonenal), are instrumental for organelle membrane permeabilization (LMP). LMP results in cation release in the cytosol, along with the activation of lysosomal degrading enzymes. These events are critical for neuronal and astrocyte death when exposed to oxidative challenges (Lee and Koh, 2010; Koh et al., 2019).
Zn2+ also affects many cytosolic pathways to promote demise in the CNS cells, including activation of apoptotic/necrotic pathways, modulation of plasma membrane channels, depletion of metabolic substrates, and the induction of cytosolic oxidative enzymes. In neurons and astrocytes, the metal contributes to NADPH oxidase activation, resulting in aberrant O2– generation (Noh and Koh, 2000; Brennan et al., 2009; Swanson and Wang, 2020). Similarly, Zn2+ activates the neuronal isoform of the nitric oxide synthase (nNOS), thereby promoting increased production of nitric oxide (NO) (Kim and Koh, 2002). These two pathways converge in a process in which O2– + NO generate ONOO– (peroxynitrite), a potently neurotoxic RNS (Bossy-Wetzel et al., 2004). Of note, the Zn2+-driven ROS/RNS production promotes further metal release from intracellular redox-sensitive stores (like MTs), thereby exacerbating a vicious feed-forward loop of cation dyshomeostasis (Corona et al., 2011; Slepchenko et al., 2017). At the cytosolic level, Zn2+ promotes NAD+ depletion, thereby resulting in glyceraldehyde-3-phosphate dehydrogenase (GAPDH, a critical enzyme in the glycolytic pathway) inhibition, ATP breakdown, and eventually energetic neuronal failures (Sheline et al., 2000). This NAD+ depletion may critically impinge on mechanisms that are relevant to aging (Lautrup et al., 2019). Zn2+ also targets and promotes activation of PARP (Kim and Koh, 2002), cyclin-dependent kinase 5 (Cdk5; Tuo et al., 2018), and AMPK (Kim et al., 2020), three molecules involved in cell death pathways.
Finally, Zn2+ mobilization, by contributing to the activation of the CamKII/p38/syntaxin/calcineurin axis, promotes outward potassium (K+) currents, a critical step in the production of neuronal apoptosis (Yu et al., 1997; McCord and Aizenman, 2013; Shah and Aizenman, 2014; Aizenman et al., 2020).
The Resilience of nNOS (+) Neurons: a Model to Investigate Excitotoxic Mechanisms
Intriguingly, some neuronal subpopulations are mostly insensitive to excitotoxicity. The phenomenon is present in oculomotor neurons, Onuf’s nucleus neurons, and NADPH-diaphorase neurons (Koh et al., 1986; Brockington et al., 2013). NADPH-diaphorase neurons are a subset of medium-sized aspiny interneurons that are largely spared following excitotoxic hits (Koh et al., 1986; Koh and Choi, 1988; Uemura et al., 1990; Weiss et al., 1994; Granzotto and Sensi, 2015). The subpopulation is characterized by the overexpression of nNOS [also known as NOS1; hereafter termed nNOS (+) neurons (Dawson et al., 1991; Hope et al., 1991)]. nNOS (+) neurons are present with a relatively more significant percentage in the striatum but are also expressed in good numbers in the hippocampus and the cerebral cortex. The subpopulation encompasses various cellular subtypes characterized by distinct morphological, transcriptomic, and functional features (Tricoire and Vitalis, 2012). Early studies have shown that these neurons survive instead of the widespread neuronal loss documented by brain autopsy of AD, HD, and PD patients, three conditions characterized by a robust glutamatergic overdrive (Ferrante et al., 1985; Graveland et al., 1985; Mufson and Brandabur, 1994).
Our group has recently exploited this neuronal subpopulation’s unique features to evaluate the mechanisms that promote resilience to excitotoxicity. Employing an array of single-cell imaging and biochemical approaches, we have demonstrated that nNOS (+) neurons fail to generate ROS in response to excitotoxic stimuli (Canzoniero et al., 2013; Granzotto and Sensi, 2015), a critical step that contributes to their resilience and enhanced survival upon glutamate-driven neurodegeneration.
The investigation of these processes has indicated an intriguing scenario in which the ROS-dependent release of intracellular Zn2+ acts as a critical intermediate step of the excitotoxic process (Granzotto and Sensi, 2015). Thus, experimental data support the notion that Zn2+ participates, with glutamate and Ca2+, in a neurotoxic ménage-à-trois.
Overactivation of NMDARs is the first mandatory step in the excitotoxic cascade; compelling evidence indicates that the receptor triggers the activation of early signaling pathways involving PSD95 and nNOS recruitment as well as aberrant Ca2+-driven induction of nNOS (Szydlowska and Tymianski, 2010; Fricker et al., 2018; Wu and Tymianski, 2018). Disruption of the NMDAR/PSD95/nNOS axis prevents excitotoxic damage in in vitro and in vivo preclinical models of cerebral ischemia (Aarts et al., 2002). Functional, transcriptomic, and biochemical analysis, however, indicate that nNOS (+) neurons express fully operational NMDARs that do not differ from the ones present in the general population of nNOS (−) neurons (Price et al., 1993; Landwehrmeyer et al., 1995; Canzoniero et al., 2013; Granzotto and Sensi, 2015; Granzotto and Sensi, 2015, observations). Interestingly, additional studies have also indicated that nNOS (+) neurons are positive to cobalt staining, a maneuver employed to identify CP-AMPARs, thereby suggesting that these cells possess a significant number of these glutamate receptor subtypes (Weiss et al., 1994). This set of findings supports the notion that NMDAR-driven Ca2+ overload and nNOS activation are necessary but not sufficient steps for the initiation and development of the excitotoxic cascade. Additional downstream processes are required, and Zn2+ participates in these mechanisms with a leading role.
Mitochondria, the Final Common Pathway
Early studies indicated that mitochondria are critical hubs for the development of the excitotoxic cascade (Ankarcrona et al., 1995). The organelles participate in the clearance of NMDAR-driven cytosolic Ca2+ raises and are instrumental for the activation of apoptotic and necrotic processes (Ankarcrona et al., 1995; Schinder et al., 1996). Mitochondrial Ca2+ overload results in organelle dysfunction, aberrant ROS generation, and, ultimately, neuronal loss (Dugan et al., 1995; Stout et al., 1998; Duchen, 2012; Rizzuto et al., 2012).
Mitochondria of nNOS (+) cells are insensitive to excitotoxicity and have emerged as a critical switch to turn off the injurious process (Canzoniero et al., 2013; Granzotto and Sensi, 2015; Figure 1). Although, mitochondria of these neurons take up large amounts of Ca2+, the organelles respond with minimal Δp losses and negligible generation of ROS (Canzoniero et al., 2013; Granzotto and Sensi, 2015). Early studies have shown that, to counteract the detrimental effects linked to peroxynitrite generation, nNOS (+) neurons express high levels of SOD2, the ROS quenching enzyme that is strategically localized inside of mitochondria (Gonzalez-Zulueta et al., 1998). Therefore, it is conceivable that this constitutive overexpression of SOD2 makes the subpopulation better equipped to cope with the oxidative surge produced by the excitotoxic challenges.
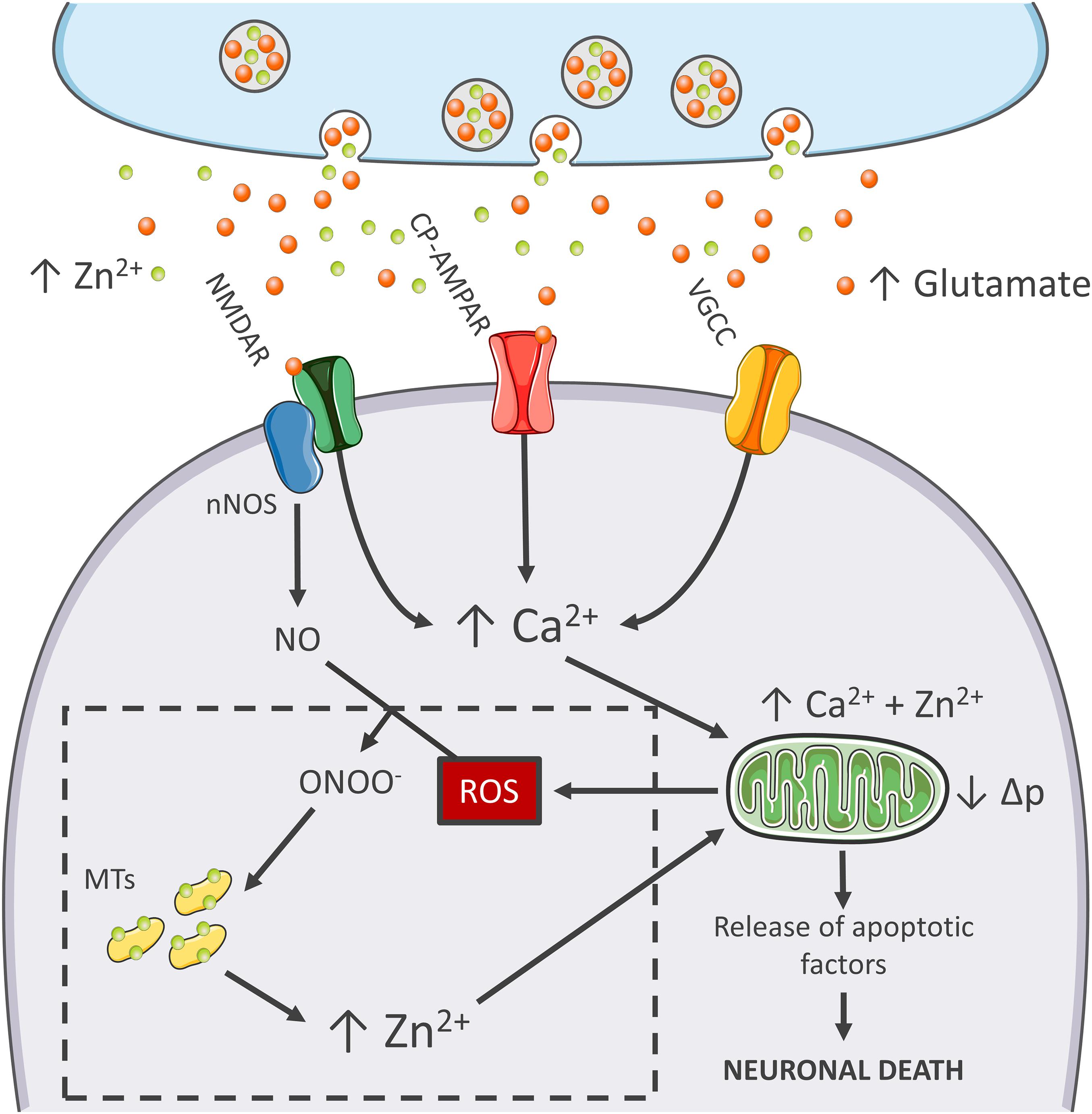
Figure 1. Zn2+ in the excitotoxic cascade. Aberrant release of glutamate from presynaptic terminals triggers NMDAR activation, which, in turn, promotes Ca2+ entry and generation of RNS and ROS of mitochondrial and extramitochondrial origin. The surge of ROS and RNS is required for intraneuronal Zn2+ mobilization from metallothioneins (MTs; Zn2+ buffering redox-sensitive proteins prone to release Zn2+ following oxidative stimuli). Intraneuronal Zn2+ rises target mitochondria and, along with Ca2+, contribute to the organelle impairment. Dysfunctional mitochondria fail to cope with Ca2+ clearance and further exacerbate Ca2+ dysregulation and ROS production. The lack of ROS generation in nNOS (+) neurons is a critical point of divergence in the excitotoxic cascade. By missing the injurious interaction between ROS and RNS, the subpopulation fails to mobilize neurotoxic Zn2+, prevents mitochondrial failure, and eventually neuronal death (dashed line box). In the general population of nNOS (−) neurons, pharmacological Zn2+ chelation prevents the full development of the excitotoxic cascade and mimics nNOS (+) cells’ behavior. These findings lend support to the idea that intraneuronal Zn2+ release is a critical regulator of excitotoxicity.
The idea that mitochondrial dysfunction and oxidative stress are prerequisites for NMDA-driven neuronal loss is in line with the “source-specific” hypothesis of excitotoxicity. The construct posits that the neurotoxic cascade depends on the route of Ca2+ entry, mainly NMDARs, rather than the magnitude of cation load (Wu and Tymianski, 2018). In agreement with this view, abundant Ca2+ entry through VGCCs, a maneuver devoid of neurotoxic effects, fails to trigger ROS and Δp changes (Table 1). This phenomenon shows great analogies with the effects of Ca2+ rises observed in nNOS (+) neurons following NMDAR activation (Granzotto and Sensi, 2015). Although, NMDAR and VGCC activation produces large Ca2+ rises, differences can be found when dissecting the temporal progression of the two stimuli. Unlike VGCC-driven Ca2+ entry, NMDAR overactivation promotes a prolonged and sustained build-up of Ca2+, a phenomenon likely due to impaired cation handling. Conceivably, the NMDAR-driven generation of RNS and ROS can severely affect the mitochondrial Ca2+ buffering as well as the defective extrusion of the cation.

Table 1. The functional hallmarks of excitotoxicity.
RNS/ROS can per se contribute to mitochondrial damage (Murphy, 2009). However, an alternative angle is offered by the mitotoxic properties of Zn2+. The cation represents a critical point of convergence between Ca2+, ROS, and mitochondrial failure. By missing ROS generation, nNOS (+) neurons fail to mobilize intracellular Zn2+ upon NMDAR overactivation (Granzotto and Sensi, 2015). Chelation experiments support the hypothesis that NMDAR-triggered Zn2+ rises are required for the full development of the excitotoxic cascade. In that respect, chelation prevents Zn2+ rises without affecting the upstream mechanisms of the cascade (i.e., Ca2+ entry or the Ca2+-driven generation of RNS/ROS). In nNOS (−) neurons, blockade of excitotoxic-driven Zn2+ elevations results in reduced mitochondrial dysfunction and improved intracellular Ca2+ cycling; two functional changes that closely match the ones observed in the nNOS (+) subpopulation (Granzotto and Sensi, 2015) and provide neuroprotection in excitotoxic settings (Wang and Thayer, 2002). On the contrary, nNOS (+) neurons are extremely vulnerable to pharmacological maneuvers that promote Zn2+ elevations, thereby suggesting that the damaging effects of the cation can override the protective machinery of nNOS (+) neurons (Granzotto and Sensi, 2015, and unpublished observations).
These results are in line with several studies showing that Zn2+ chelation is highly neuroprotective as the maneuver prevents mitochondrial failure, irreversible dysregulation of Ca2+ homeostasis, and eventually neuronal demise (Jiang et al., 2001; Bossy-Wetzel et al., 2004; Medvedeva et al., 2009; Vander Jagt et al., 2009; Clausen et al., 2013; Medvedeva and Weiss, 2014; Ji and Weiss, 2018; Ji et al., 2020; Table 1).
The peculiar milieu offered by nNOS (+) neurons replicates these mechanisms in a naïve, patho-physiologically relevant setting and allows inference on the central role played by Zn2+ in the excitotoxic process (Figure 1). Zn2+ is, therefore, not an accomplice or an amplifier of Ca2+-driven toxicity but rather the downstream executioner. Zn2+ actively promotes mitochondrial dysfunction, Ca2+ dysregulation and, eventually, neuronal death.
More at link.
No comments:
Post a Comment