https://www.frontiersin.org/articles/10.3389/fninf.2018.00026/full?
 Javaria Ashraf
Javaria Ashraf Jamil Ahmad
Jamil Ahmad Amjad Ali
Amjad Ali Zaheer Ul-Haq
Zaheer Ul-Haq- 1Research Center for Modeling and Simulation, National University of Sciences and Technology, Islamabad, Pakistan
- 2Atta-Ur-Rahman School of Applied Biosciences, National University of Sciences and Technology, Islamabad, Pakistan
- 3Dr. Panjwani Center for Molecular Medicine and Drug Research, International Center for Chemical Sciences, University of Karachi, Karachi, Pakistan
1. Introduction
Alzheimer's disease (AD) is a neurodegenerative disorder which has impacted nearly 44 million1 people around the world and this number is still increasing. AD is the leading cause of dementia in the old age (Ashford, 2004). Unfortunately, it is diagnosed only in one out of four people living with the disease1. Clinical characterization of AD includes memory loss and cognitive impairment which further lead to damaged behavioral activities and render a person completely dependent on an external aid (Budson and Price, 2005). AD establishes over time with the appearance of pathological emblems which are senile plaques and neurofibrillary tangles. These lesions comprise of extracellular deposits of Amyloid beta (Aβ) (Selkoe, 2000; Golde, 2005; Tam and Pasternak, 2012) and intracellular self-gathered clumps of tau proteins (Lee et al., 2001), respectively. Aβ is a 40–42 amino-acids long peptide which is formed after the proteolytic cleavage of Amyloid Precursor Protein (APP) (Selkoe, 2000; Golde, 2005; Tam and Pasternak, 2012). Previous studies have shown that Aβ monomers are initially non-toxic but their conversion to oligomers makes them toxic (Volles and Lansbury, 2002; Walsh and Selkoe, 2004). Eventually, the abnormal accumulation of oligomers form plaques (Walsh et al., 2002) that deposit into neuronal Endoplasmic Reticulum (ER) (Cuello, 2005) and in extracellular space (Trojanowski and Lee, 2000; Walsh et al., 2000). Aggregation of senile plaques and neurofibrillary tangles cause neuronal cell death and synaptic failure (Tiraboschi et al., 2000; Selkoe, 2002). During the last two decades, several lines of studies have pointed toward the imbalance between Aβ production and its clearance plays a central role in pathogenesis of AD. Since 1992, this hypothesis has earned acquiescence (Hardy and Higgins, 1992) and is known as “Amyloid cascade hypothesis (ACH)”. It suggests that Aβ and processing of APP are crucial in neuro-degeneration. In AD, aggregation of Aβ is the first step leading toward the formation of senile plaques (Hardy and Selkoe, 2002; Vassar, 2005). APP is a type1 trans-membrane protein produced in ER (Greenfield et al., 1999; Roussel et al., 2013). In neurons, production and metabolism of APP occurs rapidly which makes it a crucial element in neuro-pathogenesis (Lee et al., 2008). The main APP proteolytic processing steps occur at the cell surface and Trans-Golgi networks (TGNs). Proteolysis of APP can occur through the so-called non-amyloidogenic and amyloidogenic Pathways (Figure 1). The first step of non-amyloidogenic pathway is carried out by the enzyme alpha (α)-secretase that breaks down APP into soluble Amyloid precursor protein alpha (sAPPα) and alpha C-terminal fragment (αCTF / CTF83). The catalysis by α-secretase is imperative as it cuts APP within Aβ domain which blocks Aβ formation (Lichtenthaler, 2011). This initial step can also be driven by the beta (β)-secretase / β-site APP-cleaving enzyme (BACE), a transmembrane aspartyl protease (Vassar et al., 1999; Haass, 2004) (Figure 1), which constitute amyloidogenic pathway. BACE is a crucial enzyme, that acts as a rate limiting protein in Aβ generation. It breaks down the APP into soluble Amyloid precursor protein beta (sAPPβ) and beta C-terminal fragments (βCTF / CTF99) (Cai et al., 2001). The CTFs are intermediate products of the first step in both pathways which remain attached to the membrane and they are further cleaved by gamma (γ)-secretase (Zhang et al., 2011). In non-amyliodogenic pathway, the fragment α-CTF is cut down by γ-secretase into p38 and the Amyloid Precursor Protein Intracellular Cytoplasmic / C-terminal Domain (AICD). While in amyloidogenic pathway, γ-secretase degrades the βCTF into Aβ and AICD (O'Brien and Wong, 2011) (Figure 1).
FIGURE 1
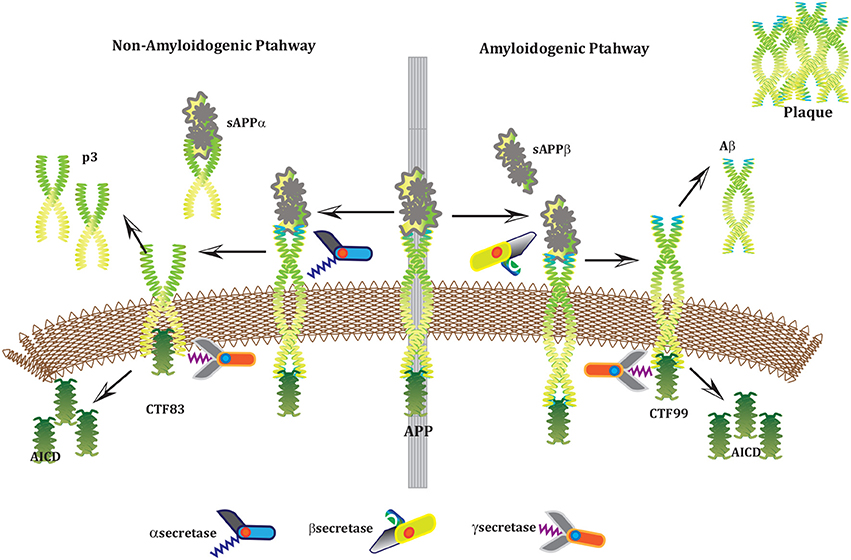 Figure 1. APP and processing products: APP is synthesized in the ER and then transported to the trans-Golgi-network (TGN) where it is cleaved by secretases. In non-amyloidogenic pathway (left), cleavage of APP by α-secretase results in the generation of sAPPα and C-terminal fragments CTF83 which is further cleaved by γ-secretase into p3 and AICD. Proteolysis by α-secretase prevents Aβ production as the cleavage site in APP is within the Aβ domain. In amyloidogenic pathway (right), APP is cleaved into sAPPβ and CTF99 by β−secretase / BACE activity. Furthermore, CTF99 breaks down into AICD and Aβ by γ-secretase activity. Aβ fragments oligomerize and fibrillize into plaques.
Figure 1. APP and processing products: APP is synthesized in the ER and then transported to the trans-Golgi-network (TGN) where it is cleaved by secretases. In non-amyloidogenic pathway (left), cleavage of APP by α-secretase results in the generation of sAPPα and C-terminal fragments CTF83 which is further cleaved by γ-secretase into p3 and AICD. Proteolysis by α-secretase prevents Aβ production as the cleavage site in APP is within the Aβ domain. In amyloidogenic pathway (right), APP is cleaved into sAPPβ and CTF99 by β−secretase / BACE activity. Furthermore, CTF99 breaks down into AICD and Aβ by γ-secretase activity. Aβ fragments oligomerize and fibrillize into plaques.
The Biological Regulatory Networks (BRN) of APP processing, depicted in Figure 2, is also built from Figure 1. APP processing depends on sequential cleavage by three secretases (α/β-secretase and γ-secretase). In normal conditions, α-secretase residing at the plasma membrane is constitutively active for APP coming to the cell surface and thus favoring non-amyloidogenic pathway (De Strooper and Annaert, 2000). Though there is an interesting fact about APP proteolysis that none of the secretases show special substrate specificity toward APP. There are several transmembrane proteins such as cell surface receptors and ligand, growth factors and cytokines besides APP which undergo ectodomain shedding by enzymes with α-secretase activity (Annaert and Saftig, 2009). In the same manner, BACE shows low affinity toward APP and it is not its exclusive physiological substrate (DeStrooper et al., 2006; Hu et al., 2006). Many observations highlight that in healthy cells APP is frequently processed through non-amyloidogenic pathway to resist amyloid generation while it is altered in pathological conditions (De Strooper and Annaert, 2000). Abnormal processing of APP is stated to be the first and fundamental step in plaques formation in AD pathogenesis (Jonsson et al., 2012). In neuropathological conditions, BACE affinity toward APP increases two folds which leads to enhanced Aβ production (Yang et al., 2003; Li and Südhof, 2004). Recent studies on transgenic mice model have shown that BACE activity is modulated by Calpain activation in AD pathology (Liang et al., 2010). Calpain-Calpastatin system also plays a key role in neurodegeneration. Transgenic mice models have shown that over expression of APP, increased production of Aβ, inhibition of Calpastatin (CAST) and activation of Calpain increase neuronal degeneration in AD (Higuchi et al., 2012).
FIGURE 2
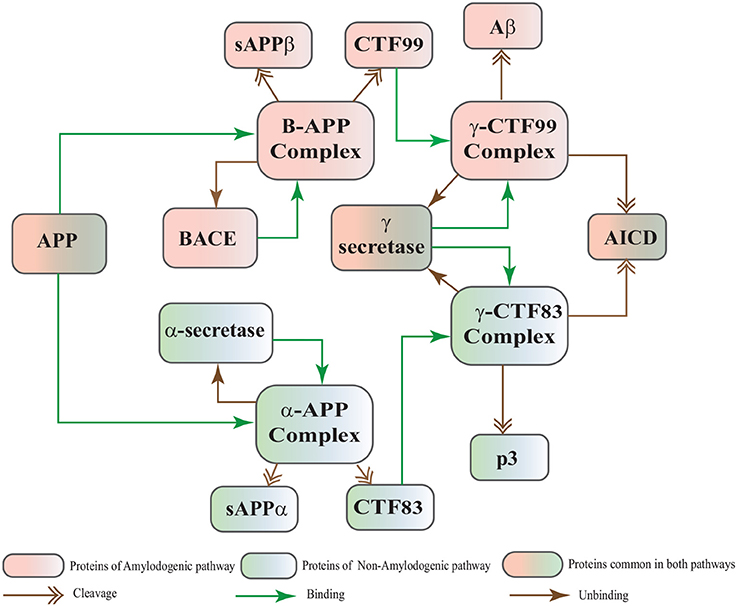 Figure 2. APP processing pathways BRN derived from Figure 1.
Figure 2. APP processing pathways BRN derived from Figure 1.
Calpains are protein clan of cysteine/ thiol proteases and their activity depends on Ca2+ concentration (Ferreira, 2012). The most studied Calpains, mu(μ)-Calpain (Calpain1) and m-Calpain (Calpain2) are present abundantly in neurons, central nervous system (CNS) and glial cells. Though their distribution differs, Calpain1 is ubiquitous and expressed more in neurons while Calpain2 is present in glial cells (Ono and Sorimachi, 2012; Santos et al., 2012). Calpain1 requires micro-molar concentration of Ca2+ (10–50μM), while Calpain2 is activated by mili-molar concentration of Ca2+ (250–350μM) in vitro (Goll et al., 2003; Ryu and Nakazawa, 2014). Ca2+ plays important role in ensuring the cell's vital functions. In addition to calcium, Calpain is tightly regulated in the cell by CAST which is also ubiquitous and solely a specific endogenous inhibitor for both Calpains (Melloni et al., 2006).
CAST is reported as an explicit suicide substrate for Calpain (Yang et al., 2013). The proportion of CAST in a cell is normally larger than Calpain, its ratio with location is crucial in controlling the extent of activation of Calpain within a cell (Todd et al., 2003). CAST interacts with Calpain at different stages i.e., first it constrains Calpain at the membrane where pro-Calpain is attached then it interacts with active Calpain inside cytosol (Hanna et al., 2008). CAST forms a reversible complex with Calpain at both the sites. At membrane, the reversible complex breaks down when Ca2+ influx increases to release Calpain. Inside cytosol, Calpain undergoes autolysis to attain active conformation. In response, CAST changes its cellular distribution to make itself widely available in the cytoplasm to counter active Calpain (Todd et al., 2003). Both active Calpain and CAST rejoin in a reversible complex to resist persistent activity of Calpain (De Tullio et al., 1999). Active Calpain modulates CAST by slowly digesting it into small inactive fragments which results in plethora of Calpain in cell leading to pathological condition (Averna et al., 2001b; Tompa et al., 2002) (Figure 3). It has been reported that in AD CAST becomes depleted from different regions of the brain as compared to healthy aged brain (Rao et al., 2008). It has also been observed that by controlling Calpain, CAST is indirectly preventing cell membrane damages induced by high Ca2+ and Aβ peptide (Vaisid et al., 2008).
FIGURE 3
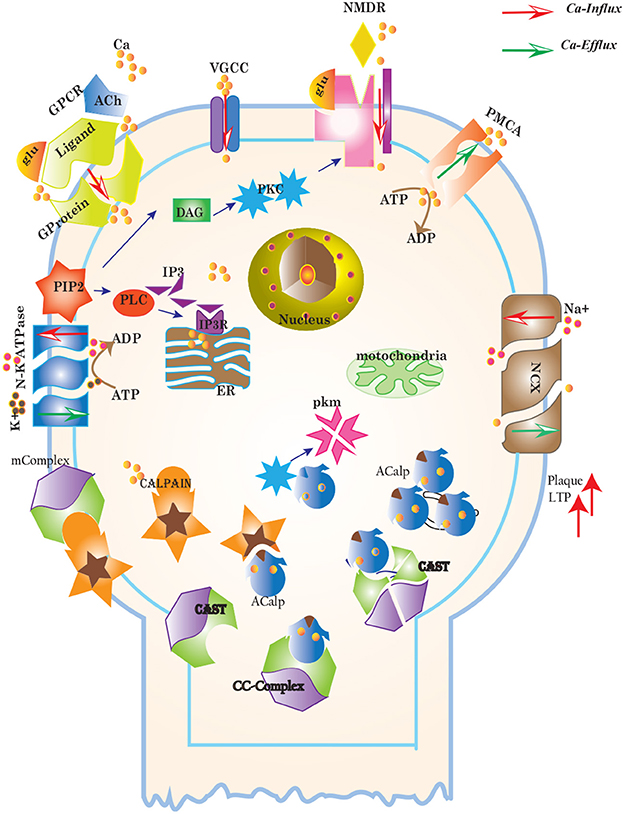 Figure 3. Different calcium channels work in harmony to establish homeostasis in neurons. Calcium influx is controlled by voltage gated (VGCCs) or receptor-ligand based (NMDAR, GPCR) channels. ER also release Ca2+ into the cytoplasm through inositol-1,4,5-trisphosphate (IP3R) and ryanodine receptors. Calcium efflux is carried out by energy (ATP) dependent channels such as plasma membrane calcium ATPase (PMCA), sodium-potassium ATPase (NKA) and sodium-calcium exchanger (NCX) channels. Calcium homeostasis influences Calpain-CAST system. At membrane, Calpain is bound to CAST to form mComplex at low Ca2+ level. At high Ca2+ concentration, Calpain is released into cytoplasm and autolysed to active form ACalp that again forms complex with CAST (cComplex). Gradually the complex breaks down and releases ACalp which enhances Plaque accumulation and LTP events.
Figure 3. Different calcium channels work in harmony to establish homeostasis in neurons. Calcium influx is controlled by voltage gated (VGCCs) or receptor-ligand based (NMDAR, GPCR) channels. ER also release Ca2+ into the cytoplasm through inositol-1,4,5-trisphosphate (IP3R) and ryanodine receptors. Calcium efflux is carried out by energy (ATP) dependent channels such as plasma membrane calcium ATPase (PMCA), sodium-potassium ATPase (NKA) and sodium-calcium exchanger (NCX) channels. Calcium homeostasis influences Calpain-CAST system. At membrane, Calpain is bound to CAST to form mComplex at low Ca2+ level. At high Ca2+ concentration, Calpain is released into cytoplasm and autolysed to active form ACalp that again forms complex with CAST (cComplex). Gradually the complex breaks down and releases ACalp which enhances Plaque accumulation and LTP events.
CAST pool is regulated by reversible phosphorylation via PKC, which is a Ca2+-activated phospholipid dependent kinase. Moreover, it is de-phosphorylated by protein phosphatases (ppase) (Melloni et al., 2006). Phosphorylation control CAST inhibitory efficiency in brain (Averna et al., 2001a) to regulate its availability for calpain inhibition. Reversible protein phosphorylation regulates many neuronal functions and is important for neuronal signal transduction (Wu and Lynch, 2006). Inactive PKC is converted to Ca2+-bound activated form in the presence of diacylglycerol (DAG) which in turn is activated by receptor based hydrolysis of phosphoinositides 3 (IP3) (Courjaret et al., 2003). The N-terminal region of CAST which is responsible for the function of the protein has a site for phosphorylation by PKC. CAST is phosphorylated by PKC to decrease its inhibitory efficiency toward calpain (Averna et al., 2001a) (Figure 3). It has been observed that PKC also regulates APP processing by activating α-secretase (Rossner et al., 2001; Racchi et al., 2003), it promotes non-amylodogenic pathway over β-secretase (Lanni et al., 2004). In vivo studies show that in the presence of PKC, secretion of sAPPα increases and Aβ secretion declines (Chen and Fernandez, 2004). Other studies about AD found that PKC has substantial role in AD pathology (Etcheberrigaray et al., 2004; Alkon et al., 2007). Active Calpain also interacts with PKC and converts it into constitutive active enzyme (Yamakawa et al., 2001; Goll et al., 2003). Calpain1 directly starts depletion of PKC from cell by converting it into protein kinase M (PKM) (Yamakawa et al., 2001; Liu et al., 2008). The whole mechanism is also depicted in the form of Calpain-CAST system BRN in Figure 4.
FIGURE 4
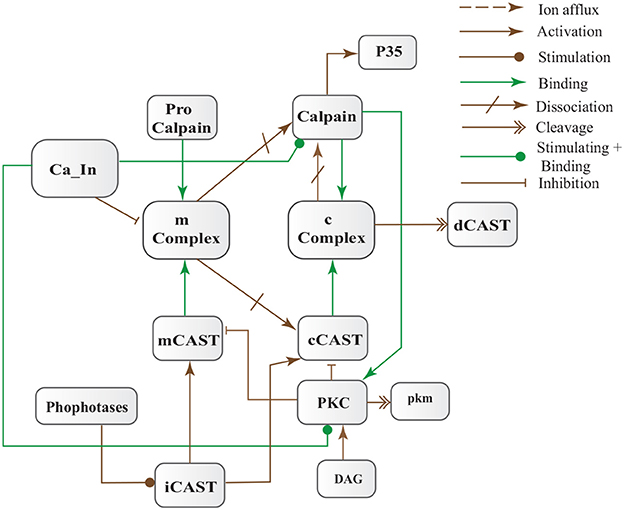 Figure 4. Calpain-CAST system BRN derived from Figure 3.
Figure 4. Calpain-CAST system BRN derived from Figure 3.
FIGURE 5
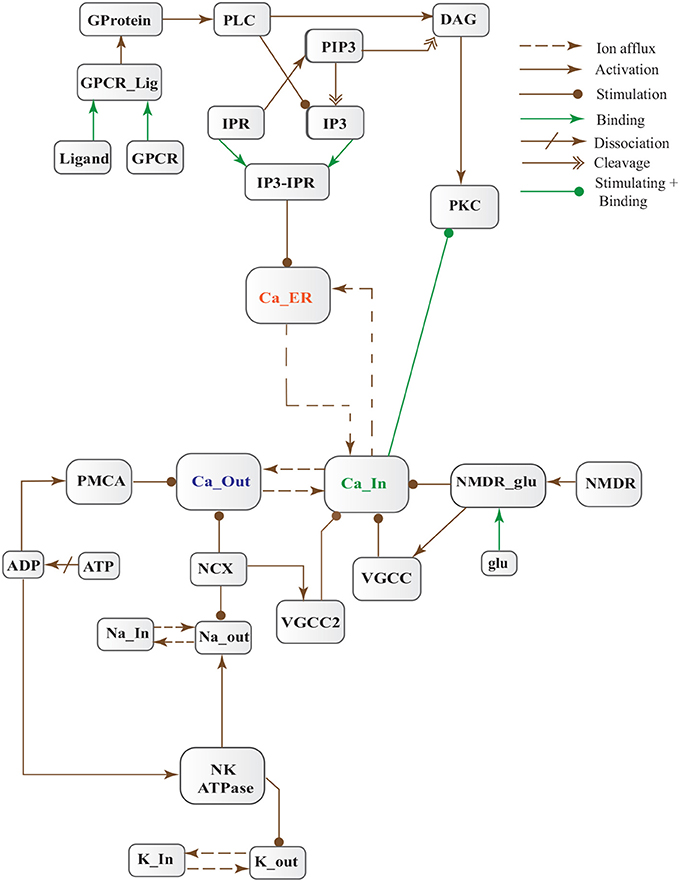 Figure 5. Calcium Influx Efflux BRN derived from Figure 3.
Figure 5. Calcium Influx Efflux BRN derived from Figure 3.
The dysregulation of Calcium homeostasis contributes in aging and neurodegeneration (Mattson, 2004; Smith et al., 2005; Stutzmann, 2005). A tremendous deal of work by calcium is tightly regulated in time, space and intensity by intracellular stores, influx and efflux channels (Stutzmann, 2005). At resting stage, extracellular Ca2+ concentration ranges from 1.5 to 2.0 mM (Orrenius et al., 2003). While magnitude of Ca2+ inside a cell is very low (between 50–100/ 50–300 nM) (LaFerla, 2002; Orrenius et al., 2003) and after activation it can rise to several micromoles. On contrary, inside ER, the level of Ca2+ is in the range 100-500μM (LaFerla, 2002) which is approximately 1000 times higher than cytosol concentration at the resting phase. Persistent alteration of Ca2+ homeostasis affects production and digestion of pathological proteins such as Calpain, Aβ and tau protein. Dysregulation of cellular Ca2+ level is an early and main feature of AD (Mattson et al., 2000; LaFerla, 2002; Small, 2009).
Cytosolic Ca2+ is maintained at very low level as compared to extracellular space through several homeostatic mechanisms, working both temporally and spatially (Figure 3). These equilibrating apparatuses include voltage-operated channels (VOCs) and receptor operated channels (ROCs) for Ca2+ inclusion, Ca2+ storage in organelles e.g., ER (Wojda et al., 2008) and Ca2+ extrusion to extracellular space. Different ATP-dependent membrane pumps such as plasma membrane calcium ATPase channel (PMCA) and sodium-calcium exchanger (NCX) which are dependent on sodium-potassium ATPase (NKA) (Wojda et al., 2008; Brittain et al., 2012) are used for Ca2+ efflux. In different physiological processes, elevation of Ca2+ is necessary to switch-on respective proteins. Ca2+ inclusion is administered by several routes such as N-methyl-D-aspartate receptor (NMDAR), an imperative type of ROCs, which switch into open conformation after binding of endogenous glutamate (glu) as ligand. Another important influx gateway is voltage gated Ca2+ channel (VGCC) which is in closed conformation when neuronal membrane is polarized (Schmolesky et al., 2002; Cain and Snutch, 2011). The VGCC adopts open conformation as plasma membrane depolarizes due to Ca2+/ sodium (Na+) influx through ROCs or ion channels (Weber, 2012). Ca2+ influx also increases from intracellular stores in ER through store-operated channels. There are two calcium channels in ER which are IP3-sensitive and ryanodine (RyRs)-sensitive Ca2+ stores (Berridge, 2009). IP3 driven release of Ca2+ starts by binding of G-protein coupled receptor (GPCR) on plasma membrane which induces Phospholipase C (PLC) mediated cleavage of phosphatidylinositol-4,5-bisphosphate (PIP2) on cell membrane into DAG and IP3. IP3 binds to its receptor on ER membrane and stimulate Ca2+ release into the cytoplasm (Berridge, 2009; Krebs et al., 2015). Furthermore, depletion of ER stores mediate influx of extracellular Ca2+ through store-operated channels (SOCs) (Emptage et al., 2001; Weber, 2012). The mechanism for lowering Ca2+ from cell is controlled by PMCA and NCX. Both PMCA and NCX are energy dependent while, NCX is also Na+ gradient dependent (Wojda et al., 2008). The BRN of Calcium channels, Figure 4, is also helpful in understanding the mechanism underlying the Ca2+ homeostasis.
To comprehend the above mentioned neuronal pathways, models are constructed to understand their dynamics. Stochastic approaches describe the randomness of biological system accurately as compared to ordinary differential equations. In BRNs, the activation or inhibition processes take place with random time delays, therefore, stochastic modeling frameworks are more suitable for their modeling. Petri nets provide complementary approach for both qualitative and quantitative modeling and simulation of the dynamical behavior of large systems in an intuitive way (Mounts and Liebman, 1997; Tsavachidou and Liebman, 2002; Tareen and Ahmad, 2015). The study (Tsavachidou and Liebman, 2002) shows that the Petri net models predict the experimental findings which support the soundness of these models. Stochastic petri nets (SPNs) have emerged as a promising tool for modeling and analyzing BRNs in the field of molecular biology (Goss and Peccoud, 1998). The dynamic behaviors of a variety of BRNs have been studied using stochastic simulations (Mura and Csikász-Nagy, 2008; Lamprecht et al., 2011; Castaldi et al., 2012; Marwan et al., 2012).
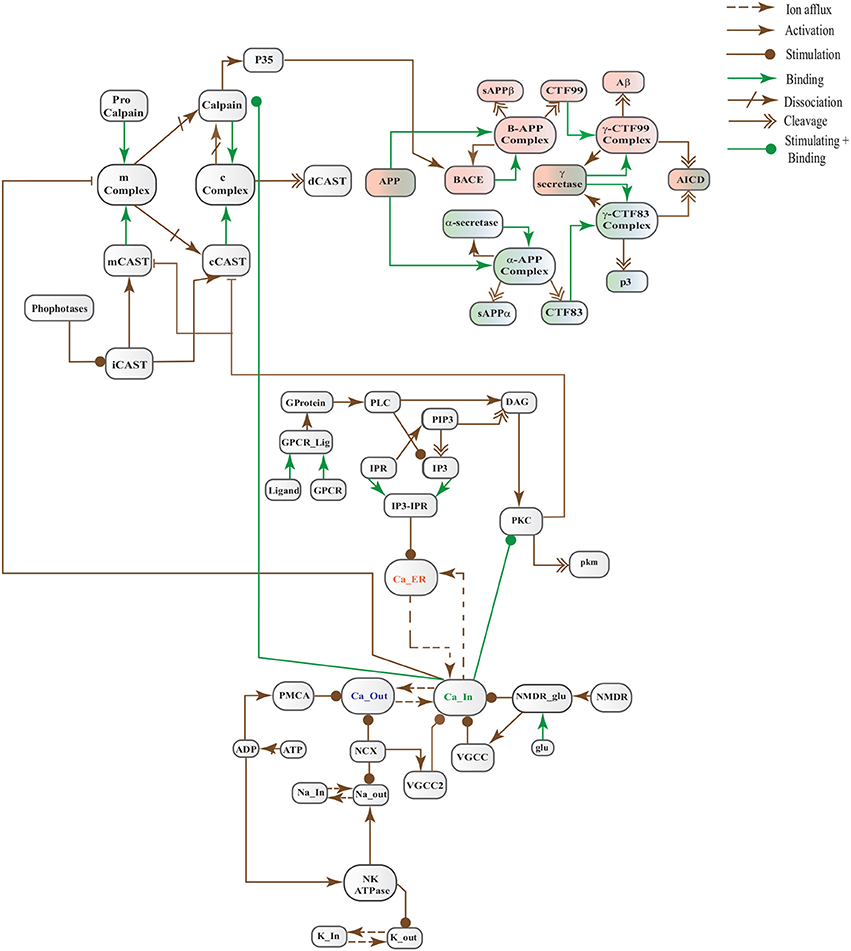
In this study, we have modeled and analyzed the neuronal physiological system constituting Ca2+ channels maintaining homeostasis, CAST regulating Calpain system and APP processing pathways separately and collectively at molecular level using SPNs to understand the AD progression mechanism. Particularly, we have analyzed neuronal patho-physiological dynamic behaviors causing the development of hallmark lesions in brain to answer many question e.g., how dysregulation of Ca2+ triggers AD? When CAST, the sole inhibitor of Calpain, depletes from the brain cells? how Aβ production increases? and when the accumulation of plaques start? The answers to these questions lie in the modeling of the combined BRN Figure 6. The model predicts that Calpain is the main cause of dysregulation, which start with the rise in Ca2+ levels in the cytosol. Calpain activates different pathways through which Aβ production and accumulation increases. Plaques start building with time at the age of forty and older. Plaques first enter lag phase and then into rapid growth phase. Calpain slowly degrades CAST which depletes from the cell and eventually neuronal degradation progresses. These results suggest that patho-physiological events such as dysregulation of Ca2+ homeostasis, Calpain hyper-activation, CAST degradation and abnormal digestion of APP, all are inter-connected and a cumulative study of these processes through SPN was needed.
FIGURE 6
No comments:
Post a Comment