I have 13 posts on reperfusion injury back to 2013, with a number of suggested interventions. I'm positive that not a single one ever made it into clinical practice, but you can always ask your doctor to try these because, 'What the hell', they might help.
Anti-Inflammatory Targets for the Treatment of Reperfusion Injury in Stroke
 Atsushi Mizuma and
Atsushi Mizuma and  Midori A. Yenari*
Midori A. Yenari*
- Department of Neurology, University of California, San Francisco and Veterans Affairs Medical Center, San Francisco, CA, United States
Introduction
Treatment of acute ischemic stroke has largely been
limited to strategies to restore blood flow. Pharmacological
recanalization, particularly tissue plasminogen activator (tPA) has been
the mainstay for acute treatment for the past 20 years (1, 2), but in recent years, several studies have shown that mechanical embolectomy is also effective (3).
However, the short time frame for safe intervention is still limited
even considering recent studies that suggest additional criteria for
treating “wake up stroke” (4) and longer time windows of up to 24 h for embolectomy (5).
It is still estimated that less than 10% of all acute stroke patients
benefit from reperfusion strategies. One of the reasons for such a short
time window is that intervention beyond this time window actually
increases risk and leads to worsened outcome (6).
If these recanalization therapies are applied too late, there is an
increased risk of cerebral hemorrhage, which can sometimes prove fatal (7).
The mechanism of cerebral hemorrhage complicating ischemic stroke is a
consequence of a phenomenon known as “reperfusion injury” (R/I) (8) to which inflammation is a major contributing cause.
While the restoration of cerebral blood flow (CBF) is a
major goal of acute stroke treatment, it can also lead to more extensive
brain tissue damage in the adjacent penumbral territory (9).
If recanalization is carried out after a critical time window, the
sudden restoration of oxygenated blood into ischemically compromised
brain tissue may overwhelm already compromised endogenous antioxidant
systems and damaged vascular endothelia leading to brain edema and
extravasation of blood cells. The generation of reactive oxygen species
(ROS) from compromised mitochondria not only leads to direct cellular
damage but also can trigger the activation of both the peripheral
(leukocytes) and brain resident (microglia) immune pathways, which in
turn, elaborate various damaging immune mediators and effectors
including more ROS. This vicious cycle in acute ischemic stroke is
referred to as cerebral R/I (Figure 1) (10, 11).
FIGURE 1
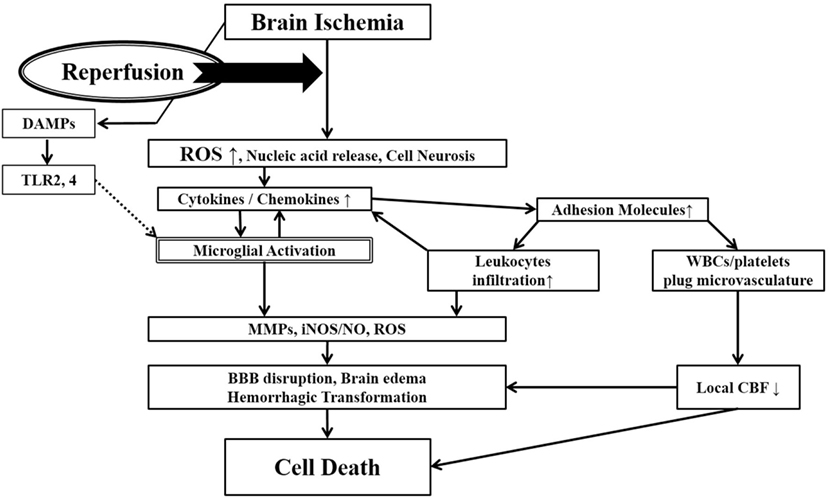 Figure 1. Ischemia-induced inflammation in
association with reperfusion injury. Once brain ischemia occurs, oxygen
and glucose supplies are reduced. If ischemia occurs for more than a
certain time period (likely a few hours, but the precise duration is not
well established) and blood flow is restored (reperfusion), worsened
injury can paradoxically occur to the brain. This is often referred to
as reperfusion injury. A major component of reperfusion injury involves
subsequent inflammatory reactions induced through various mechanisms.
Reperfusion leads to the introduction of ROS from oxygenated blood and
can stimulate an immune response in the ischemic brain. Necrotic,
ischemia-injured cells lyse and release their contents into the
extracelluar space which can act as ligands for various immune
receptors. Among these include nucleic acids which are one of many
described damage-associated molecular pattern (DAMPs, see text for
details). DAMPs can then bind TLRs and stimulate several inflammatory
responses (microglial activation, overexpression of proinflammatory
cytokines, chemokines) which lead to worsened brain injury. Inflammatory
signaling also causes immune cells to generate more effector molecules
such as ROS and iNOS/NO. In the periphery, cytokines and adhesion
molecules can attract circulating immune cells to the ischemic brain
where they infiltrate the damaged tissue and further amplify ischemic
injury. Some circulating immune cells and platelets can also plug the
microvasculature of the ischemic brain and cause secondary reductions in
local CBF. In addition to brain cells, these inflammatory reactions can
also cause damage to brain endothelia causing BBB disruption, edema and
hemorrhagic transformation. Thus, the restoration of CBF can cause more
extensive brain tissue damage. This vicious cycle is often called
reperfusion injury. ROS, reactive oxygen species; DAMPs,
damage-associated molecular patterns; TLR, toll-like receptor; MMPs,
matrix metalloproteinases; iNOS, inducible nitric oxide synthase; NO,
nitric oxide; BBB, blood–brain barrier; CBF, cerebral blood flow.
Figure 1. Ischemia-induced inflammation in
association with reperfusion injury. Once brain ischemia occurs, oxygen
and glucose supplies are reduced. If ischemia occurs for more than a
certain time period (likely a few hours, but the precise duration is not
well established) and blood flow is restored (reperfusion), worsened
injury can paradoxically occur to the brain. This is often referred to
as reperfusion injury. A major component of reperfusion injury involves
subsequent inflammatory reactions induced through various mechanisms.
Reperfusion leads to the introduction of ROS from oxygenated blood and
can stimulate an immune response in the ischemic brain. Necrotic,
ischemia-injured cells lyse and release their contents into the
extracelluar space which can act as ligands for various immune
receptors. Among these include nucleic acids which are one of many
described damage-associated molecular pattern (DAMPs, see text for
details). DAMPs can then bind TLRs and stimulate several inflammatory
responses (microglial activation, overexpression of proinflammatory
cytokines, chemokines) which lead to worsened brain injury. Inflammatory
signaling also causes immune cells to generate more effector molecules
such as ROS and iNOS/NO. In the periphery, cytokines and adhesion
molecules can attract circulating immune cells to the ischemic brain
where they infiltrate the damaged tissue and further amplify ischemic
injury. Some circulating immune cells and platelets can also plug the
microvasculature of the ischemic brain and cause secondary reductions in
local CBF. In addition to brain cells, these inflammatory reactions can
also cause damage to brain endothelia causing BBB disruption, edema and
hemorrhagic transformation. Thus, the restoration of CBF can cause more
extensive brain tissue damage. This vicious cycle is often called
reperfusion injury. ROS, reactive oxygen species; DAMPs,
damage-associated molecular patterns; TLR, toll-like receptor; MMPs,
matrix metalloproteinases; iNOS, inducible nitric oxide synthase; NO,
nitric oxide; BBB, blood–brain barrier; CBF, cerebral blood flow.
The evidence for R/I was
previously demonstrated using experimental stroke models. A few groups
have reported that ischemic injury is greater in animals where
reperfusion occurs [temporary middle cerebral artery occlusion (tMCAO)
for 2–3 h] compared to animals where there is no reperfusion (pMCAO) (12, 13).
In a series of experiments where the duration of MCAO was varied and
compared to pMCAO, tMCAO for less than 2 h led to smaller infarct sizes
compared to pMCAO (14).
Occlusion durations of more than 2 h led to paradoxically larger
infarct volumes. However, direct evidence for R/I is less clear at the
clinical level. While a rare “hyperperfusion syndrome” of accelerated
brain edema and transient clinical worsening following abrupt
revascularization has been described (15),
it is not clear whether this results in permanent worsened outcome.
Further, the neurotoxicity of tPA has been shown in previous studies,
where endogenous tPA may directly contribute to worsened outcome (16).
Further, it is quite clear that revascularization after certain time
windows can worsen outcomes compared to no intervention (6)
and could be said to represent R/I in humans. Hence, targeting aspects
of R/I might suggest an opportunity to synergistically improve
neurological outcome for thrombolysis and/or mechanical embolectomy.
While the concept of R/I as a therapeutic target
surrounding revascularization, the efficacy of treatment in experimental
reperfusion models does not necessarily predict the results of clinical
trials. A PubMed search for experimental studies covering the terms
“reperfusion injury, cerebral ischemia, and inflammation” revealed that
888 studies have been performed using the tMCAO model. However, only one
agent (edaravone) has actually been transition to the clinical level in
Japan. Experimental reperfusion models do not fully replicate what
happens in clinical stroke. Hence some reports argue that experimental
reperfusion models were inappropriate for clinical translation (17).
Regardless, the timing of treatment is different for each clinical
case. These factors are major problems that cannot be avoided. However,
some novel mechanisms associated with R/I have been established in over
the years by studying experimental models and may suggest therapeutic
targets which could be studied at the clinical level in this new era of
recanalization.
In this review, we will focus on the mechanism of R/I in
acute ischemic stroke and reconsider its treatment, with a focus on
proinflammatory targets, including some already in use at the clinical
level.
More at link.
More at link.
No comments:
Post a Comment