Use the labels in the right column to find what you want. Or you can go thru them one by one, there are only 33,532 posts. Searching is done in the search box in upper left corner. I blog on anything to do with stroke. DO NOT DO ANYTHING SUGGESTED HERE AS I AM NOT MEDICALLY TRAINED, YOUR DOCTOR IS, LISTEN TO THEM. BUT I BET THEY DON'T KNOW HOW TO GET YOU 100% RECOVERED. I DON'T EITHER BUT HAVE PLENTY OF QUESTIONS FOR YOUR DOCTOR TO ANSWER.
Changing stroke rehab and research worldwide now.Time is Brain!trillions and trillions of neuronsthatDIEeach day because there areNOeffective hyperacute therapies besides tPA(only 12% effective). I have 523 posts on hyperacute therapy, enough for researchers to spend decades proving them out. These are my personal ideas and blog on stroke rehabilitation and stroke research. Do not attempt any of these without checking with your medical provider. Unless you join me in agitating, when you need these therapies they won't be there.
What this blog is for:
My blog is not to help survivors recover, it is to have the 10 million yearly stroke survivors light fires underneath their doctors, stroke hospitals and stroke researchers to get stroke solved. 100% recovery. The stroke medical world is completely failing at that goal, they don't even have it as a goal. Shortly after getting out of the hospital and getting NO information on the process or protocols of stroke rehabilitation and recovery I started searching on the internet and found that no other survivor received useful information. This is an attempt to cover all stroke rehabilitation information that should be readily available to survivors so they can talk with informed knowledge to their medical staff. It lays out what needs to be done to get stroke survivors closer to 100% recovery. It's quite disgusting that this information is not available from every stroke association and doctors group.
Saturday, May 2, 2020
Risk factor for Alzheimer’s disease breaks the blood–brain barrier
So maybe just maybe you want your doctor to test for this APOE4 gene.
So you can kick up the Alzheimer's prevention protocol into high gear. Oh, your doctor doesn't have a protocol, too bad.
You can't use mine, I'm not medically trained, your doctors' better be EXACT.
The best-known hallmarks of Alzheimer’s
disease are clumps of misfolded amyloid-β (Aβ) and tau proteins, which
aggregate in the brain. However, there is increasing awareness that Aβ
and tau might not be the whole story — alterations in the blood–brain
barrier (BBB) have also emerged as early markers of this
neurodegenerative disorder1. The degree of disruption to the BBB correlates with the degree of cognitive dysfunction that a person experiences2, but what causes BBB breakdown has been unknown. Writing in Nature, Montagne et al.3 present evidence that the leading genetic risk factor for Alzheimer’s disease, apolipoprotein E4, is linked to BBB breakdown.
The gene apolipoprotein E (APOE) encodes a major lipid-carrier protein, ApoE, in the brain4. There are three predominant variants of APOE: APOE2, APOE3 and APOE4. As with almost all genes, people carry two copies of APOE, which can be either the same or different variants. Compared with the more-common APOE3 variant, APOE4
markedly increases the risk of Alzheimer’s disease — up to 4-fold in
people with one copy of this variant, and 15-fold in people who have two
copies4. People carrying APOE4
who do contract Alzheimer’s disease also tend to develop symptoms of
the disorder earlier than those who develop the disease but do not carry
the variant4.
Proteins
from blood plasma have been found in the cerebrospinal fluid (the
liquid that surrounds the brain and spinal cord) of cognitively healthy
people who carry APOE4 and who subsequently go on to develop
Alzheimer’s disease. These proteins have presumably leaked through the
BBB, indicating that the integrity of the barrier is lost before
cognition declines5.
Evidence from mouse models, and from the brains of people who have died
with Alzheimer’s disease, suggests that BBB breakdown is caused by the
degeneration of pericytes — cells nestled in the wall of cerebral
capillaries. These cells normally safeguard the BBB5 by preventing the breakdown of junctions between endothelial cells, which make up the capillary walls.
Whether
ApoE4 is responsible for early BBB dysfunction in Alzheimer’s disease,
by itself or in concert with Aβ and tau, was unknown. Montagne and
colleagues set out to address this knowledge gap. The authors used a
technique called dynamic contrast-enhanced magnetic resonance imaging to
investigate the permeability of the BBB in people who had either
healthy cognition or mild cognitive impairment (a prelude to Alzheimer’s
disease), grouped according to their APOE status. They found that people who were cognitively healthy and carried either one or two copies of APOE4
had a leaky BBB in two brain regions important for memory and cognition
— the hippocampus and the parahippocampal gyrus. This leakage was worse
in APOE4 carriers who exhibited mild cognitive decline.
Remarkably,
these effects preceded any signs of tissue loss in the hippocampus and
parahippocampal gyrus, attesting to the idea that BBB disruption is an
early event in the onset of neurodegeneration. BBB leakage was
independent of Aβ and tau accumulation, which the authors assessed both
by studying samples of cerebrospinal fluid and through another brain-imaging technique, positron emission tomography. Montagne and co-workers found that, unlike in APOE4 carriers, the BBB was intact in cognitively healthy APOE3 carriers. It was, however, leaky in APOE3 carriers who showed cognitive impairment — although less so than in APOE4 carriers at an equivalent stage of impairment.
Next, Montagne et al. examined whether BBB breakdown in APOE4
carriers was linked to pericyte degeneration. In support of this idea,
they found that a biomarker of pericyte injury — a soluble form of a
protein known as platelet-derived growth factor-receptor-β (sPDGFRβ) —
was elevated in the cerebrospinal fluid of APOE4 carriers compared with APOE3 carriers. High levels of the protein in people who carried APOE4 were associated with a leaky BBB and cognitive impairment. sPDGFRβ elevation was independent of Aβ and tau.
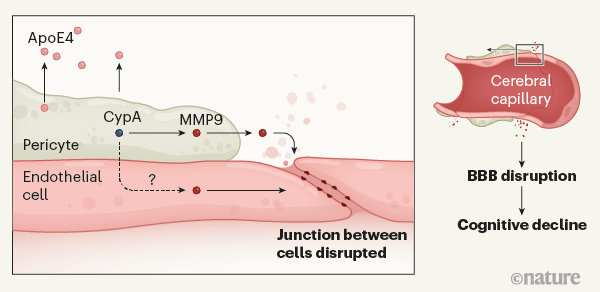
The
authors then looked for insight into the mechanisms by which pericytes
might become injured. They focused on cyclophilin A (CypA) and matrix
metalloproteinase-9 (MMP9), two proteins that are part of an
inflammatory pathway implicated in APOE4-driven pericyte damage and BBB breakdown6. Levels of CypA and MMP9 in the cerebrospinal fluid were higher in APOE4 carriers who had mild cognitive impairment than in cognitively healthy APOE4 carriers or APOE3 carriers who had comparable cognitive dysfunction. Again, this change was not related to increases in Aβ or tau.
Finally, the researchers generated pericytes in vitro from human induced pluripotent stem cells that expressed APOE3 or APOE4. They found that APOE4-expressing pericytes secreted substantially more CypA and MMP9 than did APOE3
pericytes. ApoE4 (but not ApoE3) secreted by pericytes activates the
CypA–MMP9 pathway on nearby pericytes — the cells therefore cause their
own demise. ApoE4 could also activate the CypA–MMP9 pathway in
endothelial cells, which are susceptible to the harmful effects of APOE47. Therefore, injury to pericytes and endothelial cells might both cause BBB leakage (Fig. 1).
Figure 1 | The gene variantAPOE4and Alzheimer’s disease. People who carry APOE4 are at heightened risk of Alzheimer’s disease. Montagne et al.3
provide evidence that ApoE4 protein is secreted by cells called
pericytes, which abut endothelial cells that line cerebral capillaries
at the blood–brain barrier (BBB). Secreted ApoE4 activates the protein
cyclophilin A (CypA) in the pericytes. This triggers a downstream
signalling pathway involving activation of the inflammatory protein
matrix metalloproteinase-9 (MMP9) in pericytes, and possibly also in
endothelial cells. This causes disruption of junctions between adjoining
endothelial cells, opening the BBB in brain regions involved in
learning and memory. Disruption of the BBB is associated with impaired
cognition, although the mechanisms that link the two are unclear.
These observations cast new light on APOE4 that runs
contrary to the widely held idea that this gene variant contributes to
Alzheimer’s disease solely by promoting Aβ and tau accumulation4. Instead, it seems that BBB dysfunction might explain why APOE4 carriers are susceptible to Alzheimer’s disease. The authors’ findings might also explain why APOE4 carriers have worse outcomes following stroke or traumatic brain injury8 than do people who carry other APOE variants. However, as Alzheimer’s disease progresses, APOE4 could also slow Aβ and tau clearance, exacerbating declines in cognition.
Even more striking is the finding that early drivers of cognitive impairment differ between APOE4 and APOE3
carriers. Montagne and colleagues’ findings indicate that activation of
the CypA pathway and pericyte damage might not be involved in cognitive
impairment in people who carry the most common APOE variant, APOE3.
But whether a leaky BBB caused by factors that are independent of
pericytes (for example, damage to endothelial cells caused by Aβ1) contributes to cognitive impairment in APOE3 carriers remains unclear. The role of the BBB in APOE2 carriers, which was not assessed in the current study, also remains unknown. Although APOE2 is associated with a reduced risk of Alzheimer’s disease compared with other APOE variants, this is unlikely to result from a more resilient BBB, because APOE2 carriers have an increased risk of microhaemorrhages, suggesting vascular frailty4.
Whether
and how BBB breakdown leads to cognitive impairment also remains to be
determined. Is it a cause or a consequence of the disease process?
Evidence from mice indicates that some proteins in the blood, such as
fibrinogen, damage the synaptic connections between neurons9. But a pathogenic role for these proteins in the human brain has not yet been demonstrated.
Irrespective of these questions, Montagne et al. have broadened our understanding of how APOE4 promotes cognitive impairment. They have also demonstrated that different APOE
statuses can promote disease through different mechanisms. A deeper
appreciation of how gene variants shape Alzheimer’s disease might prove
crucial for more-personalized approaches to treating this prevalent and
incurable disease.

No comments:
Post a Comment