Sounds like it might be useful in stopping this cause of the neuronal cascade of death;
Inflammatory action leaking through the blood brain barrier.
What is your doctor and stroke hospital doing to get this tested in humans for stroke?
Do you prefer your doctor and hospital incompetence in this NOT KNOWING? OR NOT DOING?
Don't do this but there is carvacrol essential oil. I'm not medically trained so don't listen to me. And this was injected in 30 minutes not misted in the air so your doctor would have to apply.
Carvacrol decreases blood–brain barrier permeability post-diffuse traumatic brain injury in rats
Scientific Reports volume 13, Article number: 14546 (2023)
Abstract
Previously, we showed that Satureja Khuzestanica Jamzad essential oil (SKEO) and its major component, carvacrol (CAR), 5-isopropyl-2-methylphenol, has anti-inflammatory, anti-apoptotic, and anti-edematous properties after experimental traumatic brain injury (TBI) in rats. CAR, predominantly found in Lamiaceae family (Satureja and Oregano), is lipophilic, allowing diffusion across the blood–brain barrier (BBB). These experiments test the hypothesis that acute treatment with CAR after TBI can attenuate oxidative stress and BBB permeability associated with CAR’s anti-edematous traits. Rats were divided into six groups and injured using Marmarou weight drop: Sham, TBI, TBI + Vehicle, TBI + CAR (100 and 200 mg/kg) and CAR200-naive treated rats. Intraperitoneal injection of vehicle or CAR was administered thirty minutes after TBI induction. 24 h post-injury, brain edema, BBB permeability, BBB-related protein levels, and oxidative capacity were measured. Data showed CAR 200 mg/kg treatment decreased brain edema and prevented BBB permeability. CAR200 decreased malondialdehyde (MDA) and reactive oxygen species (ROS) and increased superoxide dismutase (SOD) and total antioxidative capacity (T-AOC), indicating the mechanism of BBB protection is, in part, through antioxidant activity. Also, CAR 200 mg/kg treatment suppressed matrix metalloproteinase-9 (MMP-9) expression and increased ZO-1, occludin, and claudin-5 levels. These data indicate that CAR can promote antioxidant activity and decrease post-injury BBB permeability, further supporting CAR as a potential early therapeutic intervention that is inexpensive and more readily available worldwide. However, more experiments are required to determine CAR’s long-term impact on TBI pathophysiology.
Introduction
Carvacrol (2-methyl-5-(1-methyl ethyl)-phenol) (CAR) is found in oils obtained from the plants of the Lamiaceae family, such as Thym, Satureja, and Origanum genera, in concentrations of 85–90%1,2. Because of its low molecular mass and lipophilic characteristics, this molecule may easily pass across the blood–brain barrier (BBB)3. CAR is generally considered a safe food additive that can be added directly to human food4,5 and possesses various beneficial effects in vitro and in vivo, including antioxidant, anticancer, antibacterial, antifungal, anti-inflammatory, and hepatoprotective properties6,7,8,9. Recent studies have shown that CAR exerts its neuroprotective effects in brain disorders by inhibiting reactive oxygen species (ROS) production and antioxidant properties10,11.
Traumatic brain injury (TBI) is divided into two phases of pathophysiological damage: primary (e.g., brain contusion, diffuse axonal injury, and hemorrhages of parenchyma or subarachnoid region) and secondary (e.g., BBB disruption, edema, herniation, ischemia, and infarction)12. Theoretically, prevention or inhibition of early secondary injury signaling cascades will attenuate persisting pathophysiology and promote improved long-term outcomes. The BBB selectively restricts the paracellular diffusion of compounds from the blood to the brain through specialized endothelial cells connected by tight junctions. Tight junctions consist of scaffolding proteins, like zonula occludens (ZO), occludins and claudin-5, that are responsible for the structural integrity of the BBB13,14. Astrocyte end-feet and microglial processes interact with the brain endothelium, forming the gliovascular unit responsible for maintaining cerebral homeostasis and optimal neuronal activity15.
In addition to axonal injury, mild-severe TBI causes mechanical depolarization and spreading depolarization, increased intracellular Ca+2 levels, and decreased cerebral blood flow, resulting in a global metabolic crisis. Consequently, increased nitric oxide (NO) synthase ROS activity offset the capabilities of endogenous antioxidants (e.g., glutathione peroxidase, superoxide dismutase (SOD)), leading to oxidative stress. Oxidative stress wreaks havoc by modulation of vascular function, triggering cell death cascades, activating enzymes (e.g., matrix metalloprotease-9 (MMP-9)), damaging nucleic acids, and oxidizing fatty acids, amino acids, and co-factors of cellular processes12,16. Along with other secondary injury cascades, oxidative stress contributes to immediate and delayed BBB permeability allowing the diffusion of blood-borne molecules into the extracellular matrix of the brain, which further promotes oxidative and inflammatory states that lead to excessive MMP-9 activity17,18 (Fig. 1).
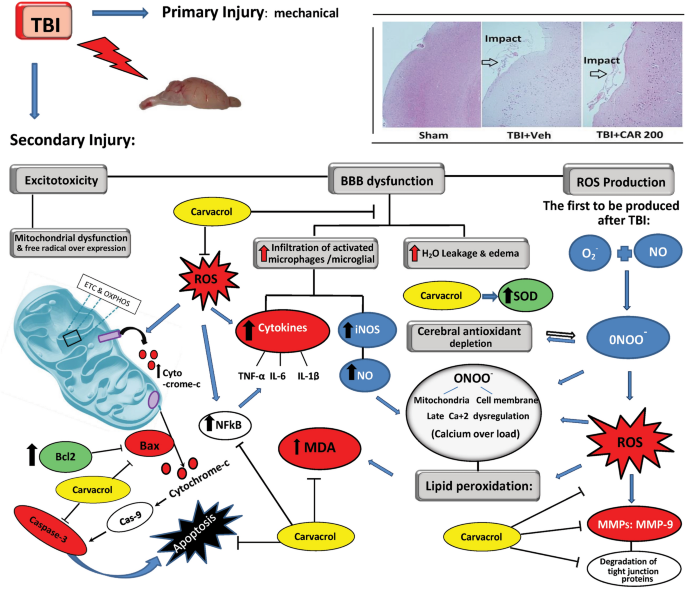
Schematic representation of the biochemical and molecular processes characterizing the TBI-mediated secondary damage. TBI induces excitotoxicity, resulting from excessive glutamate release, along with alteration of the blood–brain barrier (BBB) permeability, Malfunctioning of the mitochondrial, and free radical overexpression. TBI causes increased intracellular Ca+2 levels resulting from dysfunction of the mitochondrial electron transport chain (ETC) and oxidative phosphorylation (OXPHOS). This would lead to an increased nitric oxide (NO) synthase and reactive oxygen species (ROS) affecting endogenous antioxidants (e.g., glutathione peroxidase, superoxide dismutase; SOD) functions leading to oxidative stress. On the other hand, BBB permeability causes vasogenic brain edema and infiltration of activated macrophages/microglia resulting in (NO) production. Taking the processes together,
) generation. Peroxynitrite and ROS actively take part in lipid peroxidation, DNA damage, and protein oxidation. Oxidative stress also wreaks havoc by modulation of vascular function, triggering cell death cascades, and activating enzymes (e.g., matrix metalloprotease-9; MMP-9). Carvacrol may alleviate the aforementioned destructive mechanisms of the cell by inhibiting ROS.
MMP-9 is involved in several cellular processes, such as healing, death, and morphogenesis. After TBI, MMP (including MMP-9) activity is overexpressed, where MMP-9 mediates degradation of ZO-1, occludin, and claudin-5, responsible for increased permeability of the BBB19 (Fig. 2). In animal studies and patients with TBI, MMP-9 is implicated in the pathogenesis of cerebral edema and has been effective in predicting the outcomes of neurological disorders20,21.
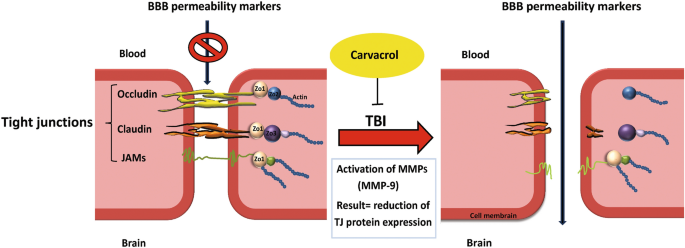
Intraperitoneal administration of Carvacrol 200 mg/kg at 30 min post-TBI suppressed over expression of MMP-9 which mediates degradation of ZO-1, occluding and claudin-5 proteins responsible for increased permeability of BBB. TBI, traumatic brain injury; BBB, blood brain-barrier.
In more severe cases of TBI, cerebral hemorrhage and edema caused by BBB destruction are among the leading causes of mortality in patients with TBI22. In TBI, brain edema is divided into cytotoxic and vasogenic edema. Vasogenic edema results from increased BBB permeability allowing extravasation of fluid and plasma into the brain and is associated with acute edema after closed head injury. Cytotoxic edema is cellular swelling typically associated with significant cellular damage and disruption of ion homeostasis. While both forms of edema occur after TBI, vasogenic edema via BBB permeability is best supported in the literature23.
Our previous study showed CAR administered 30 min post-TBI significantly decreased brain edema, neuroinflammatory markers, and NF-κB and caspase-3 mediated cell death pathways in rats24. In neurotoxic environments, CAR treatment reduces oxidative stress in the brain7,25,26; however, the antioxidant properties of CAR have not been evaluated after TBI. We hypothesize that CAR treatment will reduce oxidative stress and MMP-9 activity, thereby preventing the degradation of tight junctions and preserving BBB integrity after experimental TBI, thereby reducing edema. The efficacy of CAR to prevent or attenuate TBI-induced BBB permeability and oxidative stress would be an optimal translatable treatment strategy that could improve TBI outcomes when added to the current standard of care.
Results
CAR treatments improved veterinary coma scale (VCS) after TBI
Neurological outcomes (VCS scores) among the different groups were evaluated as a function of time post-injury and treatment group (Fig. 3). There was no significant difference in VCS scores between groups before TBI. The VCS scores were significantly decreased in the TBI groups at 4 h (9.33 ± 0.33) and 24 h (11.22 ± 0.23) in comparison with the sham group (15.0 ± 0.0; P < 0.001, respectively). However, the VCS scores were increased in the CAR200 group at 4 h (12.33 ± 0.3) and 24 h (14.5 ± 0.22) post-TBI as compared to the TBI + Veh group (P < 0.001, respectively). There was no significant difference in the VCS between the TBI, TBI + Veh and TBI + CAR100 groups. VCS scores in the CAR200 naive treated-rats group remained unchanged compared to the sham group (Table 1).
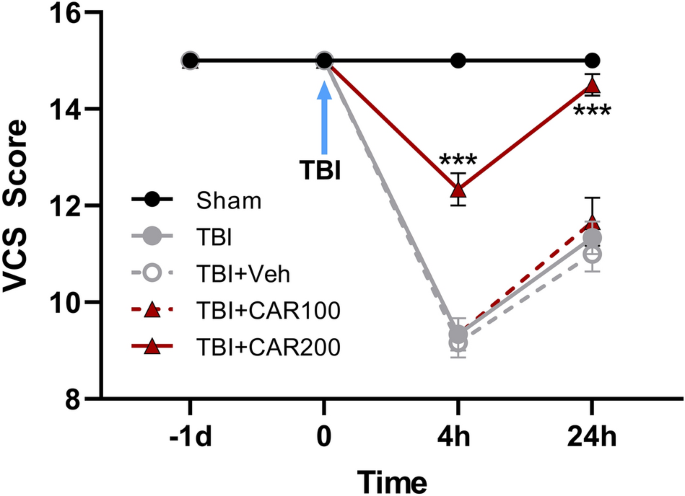
Veterinary coma scale (VCS) scores 4 and 24 h after TBI induction in the different experimental groups (n = 6 in each group). Data are expressed as mean ± SEM. ***P < 0.001 at 4 and 24 h (hr) between TBI + CAR200 and the other groups. TBI decreased the VCS scores significantly at 4 and 24 h in all the groups. However, CAR200 caused greater improvement in the VCS scores compared to the other treatments. TBI, traumatic brain injury; CAR, carvacrol.
CAR reduces TBI-induced cerebral edema
Figure 4A shows the effect of different doses of CAR on BWC. The percent BWC in the TBI (76.88 ± 0.58), TBI + Veh (76.02 ± 0.51), and the TBI + CAR100 (74.93 ± 0.73) groups were higher than that of the sham group (70.83 ± 0.23; P < 0.001). In contrast, the percent BWC in the CAR200 group (69.55 ± 0.78) was significantly reduced compared to the TBI + Veh group (P < 0.001). There was no significant difference in the BWC between the TBI and TBI + Veh groups. CAR200 did not affect BWC in naïve rats compared to the sham group (Table 1).
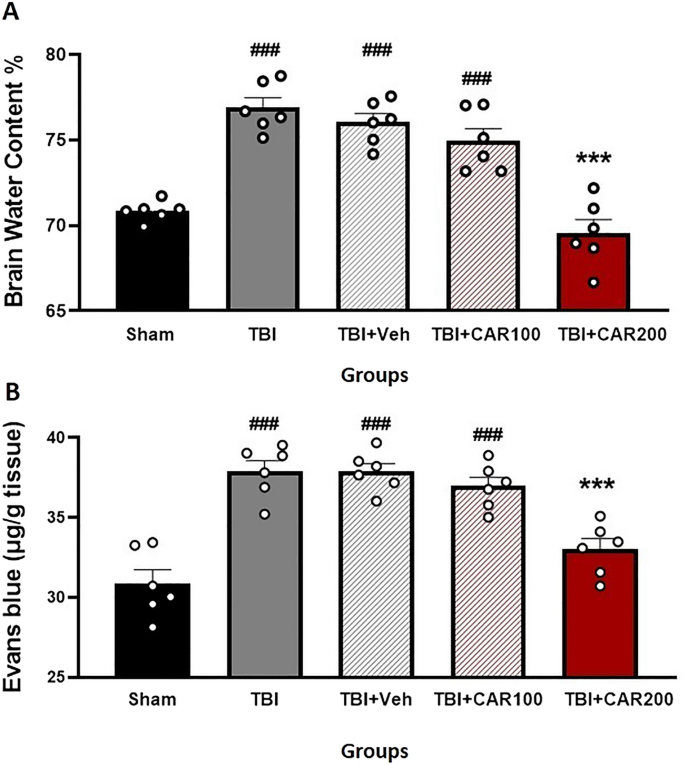
Effect of CAR (200 mg/kg i.p.) on the brain water content (BWC) (A) and the Evans blue dye content (μg/g tissue) (B) 24 h after TBI induction in the different experimental groups (n = 6 in each group). Data are expressed as mean ± SEM. ###P < 0.001 represents a significant difference with sham; ***P < 0.01 represents a significant difference with TBI + Veh. TBI, traumatic brain injury; CAR, carvacrol.
CAR decreased blood–brain barrier permeability
To clarify the vasogenic edema, brain EB dye content, a commonly used marker of brain barrier integrity and vascular permeability, was measured (Fig. 4B). The EB dye content in the TBI group was significantly increased compared to the sham group (37.95 ± 0.51 vs. 30.86 ± 0.80, P < 0.001). The TBI + CAR200 group showed a significant decrease in EB dye content compared to the TBI + Veh group (33.01 ± 0.56 vs. 37.88 ± 0.50, P < 0.001), while CAR100 was similar to the TBI + Veh group. The EB dye content was not significantly different between the TBI and TBI + Veh groups. Our data illustrate the significant potency of CAR200 on alleviating vasogenic edema.
CAR reduced TBI-induced oxidative stress
The tissue level of MDA increased by 162% in the TBI + Veh group compared to that of the sham group (12.56 ± 0.87 vs. 4.80 ± 0.24, P < 0.001). Treatment with CAR200 reduced the MDA levels by 19% compared with the TBI + Veh group (10.19 ± 0.40, P < 0.05; Fig. 5A). The level of brain ROS was significantly increased by 58% in the TBI + Veh group as compared to the sham group (166.5 ± 5.95 vs. 69.5 ± 4.17, P < 0.001). CAR200 treatment after TBI reduced the level of ROS by 36% compared with the TBI + Veh group (106.75 ± 7.46, P < 0.001; Fig. 5B). SOD activity decreased by 66% in the TBI + Veh group compared with the sham group (7.39 ± 0.23 vs. 2.53 ± 0.47, P < 0.001). Administration of CAR200 significantly increased SOD activity by 112% compared with the TBI + Veh group (5.36 ± 0.44, P < 0.01; Fig. 5C). T-AOC in the TBI + Veh group was decreased by 58% compared to the sham group (207.25 ± 34.70 vs. 492.75 ± 10.28, P < 0.001). TBI + CAR200 increased T-AOC by 68% compared to the TBI + Veh group (348.5 ± 25.52, P < 0.01; Fig. 5D).
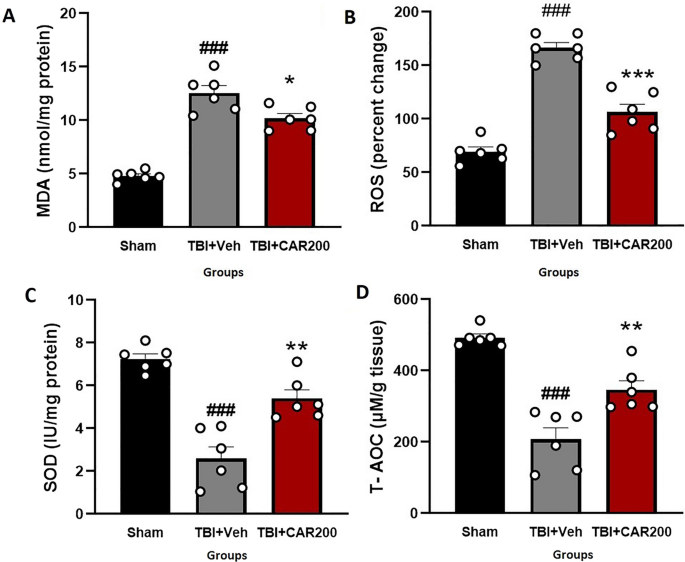
Effect of CAR (200 mg/kg i.p.) on the brain tissue MDA levels (A), ROS (B), SOD activity (C), and T-AOC (D) 24 h after TBI induction in the different experimental groups (n = 6 in each group). Data are expressed as mean ± SEM. ###P < 0.001 represent significant differences with sham; *P < 0.05. **P < 0.01 & ***P < 0.001 represent significant differences with TBI + Veh. TBI, traumatic brain injury; CAR, carvacrol; MDA, malondialdehyde; ROS, reactive oxygen species, SOD, superoxide dismutase; T-AOC, total antioxidative capacity.
CAR preserved tight junction proteins after TBI
The level of ZO-1 (Fig. 6A and C) and occludin (Fig. 6A and D) proteins were significantly reduced by 76% and 70%, respectively, in the TBI + Veh group compared to the sham group (1.0 ± 0.03; 1.0 ± 0.04, respectively) at 24 h after injury (0.24 ± 0.03; 0.30 ± 0.01, respectively; P < 0.001). CAR200 treatment significantly increased the expression levels of ZO-1 and occludin 313% and 127%, respectively, compared with the TBI + Veh group (0.99 ± 0.19; 0.68 ± 0.11, respectively). Claudin-5 decreased by 72% in the vehicle-treated injured animals (TBI + Veh group, 0.28 ± 0.05) compared to the sham group (1.0 ± 0.03). CAR200 significantly increased the level of claudin-5 by 229% in comparison with the TBI + Veh group (0.92 ± 0.08; 0.28 ± 0.05, respectively) (Fig. 6A and E).
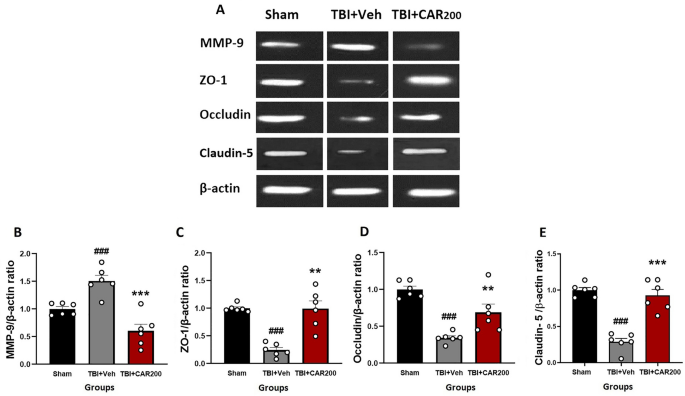
Effect of CAR (200 mg/kg i.p.) on the expression of MMP-9 (B), ZO-1 (C), occludin (D), and claudin-5 (E) 24 h after TBI induction in the different experimental groups (n = 6 in each group). Data are expressed as mean ± SEM band density ratio for each group. β-actin was used as an internal control. ###P < 0.001 represents significant difference with sham; **P < 0.01 & ***P < 0.001 represent significant differences with TBI + Veh. TBI, traumatic brain injury; CAR, carvacrol; MMP-9, matrix metalloproteinase-9, ZO-1, zonula occludens-1. Representative bands of the Western blot from the same run are shown above in (A).
CAR reduced MMP-9 protein levels after TBI
A 50% upregulation in MMP-9 level was observed in the TBI + Veh group compared to the sham group 24 h after brain injury (1.50 ± 0.02 vs. 1.0 ± 0.005, P < 0.001) (Fig. 6A and B). CAR200 administration significantly downregulates MMP-9 expression by 60% compared to the vehicle-treated group (0.60 ± 0.15, P < 0.001).
Discussion
Data support the novel hypothesis that acute treatment with CAR after TBI can attenuate BBB permeability, in part, by decreasing oxidative stress and reducing MMP-9 levels that could be responsible for the preservation of ZO-1, occludin, and claudin-5 levels at 24 h post-injury. Further, the optimal dose of CAR, 200 mg/kg, was identified. Last, the optimal dose of CAR did not cause changes in heart rate, blood pressure, or body weight. Together, these data support that CAR may be beneficial in reducing vasogenic edema as a post-TBI therapeutic intervention that warrants further investigation.
ROS are natural byproducts of oxygen metabolism. Free radicals like superoxide (O2-), hydroxyl radicals (
), which degenerates into various unstable reactive nitrogen species (RNS). Peroxynitrite, RNS, and ROS actively participate in lipid peroxidation, DNA damage, and protein oxidation32. By definition, lipid peroxidation is a free radical-mediated event, that can result in the breakdown of polyunsaturated fatty acids in lipid membranes, producing MDA33 leading to oxidative neural injury post-TBI.
Our results showed that the ROS and MDA levels were significantly higher in the TBI + Veh group compared to the sham group, similar to other groups using this experimental model of TBI34,35.Our data indicated that the percentage of ROS and MDA generation was lower in the CAR-treated group (36% and 19%; respectively) than in the vehicle-treated group, indicative of the neuroprotective role of CAR treatment. There is a growing body of evidence on the role of CAR on free radical scavenging, which has been also previously shown in in vitro and in vivo studies36. For instance, Samarghandian and colleagues administrated CAR to rats that were exposed to chronic stress. They showed that CAR not only alleviates free radicals such as peroxide, H2O2, superoxide, and NO but also improves the activity of antioxidant enzymes, such as SOD, CAT, and GPx. In their study, CAR exerted a protective effect on the hippocampus of rats via decreasing lipid peroxidation7. In addition, Wang et al.37 revealed that CAR alleviated lipid peroxidation in the hippocampus of ethanol-exposed rats. In line with the antioxidant effects of CAR, Li et al.38 have shown that CAR inhibits the oxidative response in a rat model of focal cerebral ischemia by increasing SOD activity and decreasing MDA levels. Therefore, probably part of the protective actions of CAR in our study may be related to its increasing antioxidant power (SOD & T-AOC) and reducing oxidative stress. Clinical studies also indicate the antioxidant effects of CAR39. For example, Khazdair and colleagues recently administered CAR for two months on 20 patients exposed to sulfur mustard (SM)-inducing lung disorders 27–30 years ago. CAR significantly reduced MDA, increased the SOD and CAT factors, and improved forced vital capacity and peak expiratory flow in these patients40.
Our data also showed that CAR200 treatment reduced TBI-induced injury severity by over 50% at 4 h post-injury, returning to sham levels by 24 h post-injury, demonstrating that improved oxidative homeostasis and preserved BBB could promote behavioral recovery. It is well known that the prevention of lipid peroxidation is vital to improving neurological outcomes in clinical and preclinical studies so that the improved VCS scores in the CAR-treated group may relate to a limited level of MDA41. Furthermore, we speculate that the improvement of neurological behavior in the group treated with CAR is due to the correlation of antioxidant property of this substance and apoptotic process, because excessive oxidative stress induces resulting in releasing cytochrome c, a protein that plays a crucial role in cell death and is inhibited by Bcl-2 protein, from outer mitochondria membrane (OMM). Then, cytochrome c forms the apoptosome complex in the cytosol. These two mechanisms converge at the level of effector caspases such as caspase-3 and caspase-7, resulting in cleavage of cellular proteins and apoptosis42. It has also been demonstrated that decreasing caspase-3 activation and apoptotic cell death promotes functional recovery in animals following TBI43,44. It is important to note that 24 h after administration of CAR200, in our previous study, Bax/Bcl-2 protein ratio and cleaved caspase-3 synthesis in the brains of rats decreased, and consequently neurological scores improved24. Therefore, CAR may have reduced neurological deficit post-TBI by reducing ROS and subsequent reduction in apoptosis.
Of interest, several studies have found a potent link between oxidative stress and MMP-9 in the pathophysiology of BBB damage after TBI18. Multiple cellular and molecular mechanisms that induce MMP-9 activation can be triggered by oxidative stress45. MMP-9 contributes to the degradation of tight junction proteins (ZO-1, occluding and claudin-5) affecting BBB permeability46. While ZO-1 and occludin are important regulators of the blood–brain barrier, claudin-5 plays a crucial role in maintaining barrier integrity, where claudin-5 levels have been linked to increased permeability and disruption of the blood–brain barrier47. Our findings indicate that CAR effects on BBB permeability and brain edema might be attributed to a reduction in oxidative stress resulting in the modulation of MMP-9 expression and BBB tight junction proteins. Supporting these outcomes, in-vitro studies showed the capability of CAR to decrease the MMP-9, MMP-2, and ROS in human glioblastoma and human mesenchymal stromal48,49.
One of the possible mechanisms through which CAR reduced MMP-9 could be c-Jun N-terminal protein kinase (JNK). According to recent research by Lu et al.17, the oxidative stress and JNK pathway significantly increase the magnitude of MMP-9 activation resulting in the degradation of tight junction proteins and increased BBB permeability in the rat brains after TBI. Gholijani et al.50 suggested that CAR can inhibit JNK pathways in lipopolysaccharide (LPS)-stimulated mouse macrophages. The underlying mechanisms through which CAR administration changes JNK expression might differ in various animals and cell types and has not been evaluated after TBI.
In addition to affecting the JNK pathway, ROS also has stimulating effects on the transcription of NF-κB which appear to play a major role in perpetuating the immune response to TBI and is known to increase genetic transcription of pro-inflammatory mediators such as TNFa and IL-651. It has been clarified that inflammatory cells and BBB permeability are related to neurological defects following TBI52. In this case, inhibition of NF-κB blocks MMP-9 upregulation in ischemic brain endothelium53. It has been also observed that there is a correlation between neurological scores and reducing MMP-9 mRNA and protein expression levels in patients with brain trauma21. Based on our previous work, we showed that CAR-treated animals had significantly lower pro-inflammatory cytokines and NF-κB protein post-TBI24. Thus, it is reasonable to speculate that better neuronal function following CAR treatment may be associated with BBB protection and suppression of the ROS/NF-κB/MMP-9 pathway.
Limitations. (1) Evans blue used in our study is an inexpensive and widely available transcellular marker of albumin extravasation across the blood–brain barrier, often used to determine vasogenic edema and BBB permeability after TBI. Due to the increased chance of detection in controls, it is less precise than paracellular tracer permeability, like dextran and inulin54. However, in a study by Liao et al.55, EB and dextran were compared and indicated that EB could detect BBB permeability, but non-specific signals were reduced in shams using dextrans. So, the increase in extravasation in injured animals in these experiments was likely due to BBB permeability caused by the TBI. (2) The experiments only used male rats, despite growing evidence indicating there are sex differences in the acute glial response, including astrocytes, microglia, and infiltrating macrophages that can lower the amount of MMP activation and subsequent degradation of the BBB at the 24 h and 7 days time points. However, there is support that this may be due to a delayed response after TBI, where more chronic time points reveal similar long-term pathology56,57. Based on these publications, it is likely that a sex-dependent response would be present that requires females to be included in future experiments and in the evaluation of long-term outcomes. (3) Our experiments only evaluated outcome measures at 24 h. Longitudinal evaluation of both pathophysiological sequelae and the influence on long-term neurological function would improve overall interpretation regarding any potential benefit or early intervention with CAR. (4) Our findings demonstrated specific antioxidant properties of CAR; however, CAR may also have broader neuroprotective effects beyond its antioxidant properties. For example, CAR has been shown to have anti-inflammatory effects, contributing to activated microglia and astrocytes, where reducing inflammation can help to maintain BBB integrity58. It is also possible that CAR may reduce inflammation, which in turn reduces oxidative stress59,60. Additionally, CAR may regulate other pathways that contribute to BBB integrity and neuroprotection, indicating the potential for also mediating aspects of cytotoxic edema, which is typically more associated with focal injuries. Further research is needed to fully understand the potential therapeutic applications of CAR in this context.
Conclusions
Our findings indicate that CAR prevents the loss of ZO-1, occludin, and claudin-5 proteins after TBI, through a MMP-9 signaling pathway. Our results also revealed that the impact of CAR treatment on MMP-9 might be partially attributed to a decrease in the generation of oxidants such as MDA and ROS and an increase in antioxidants like SOD activity and T-AOC. The 200 mg/kg dose of CAR did not cause any acute changes in blood pressure, heart rate, or body mass, indicating that it may be the optimal dose to evaluate the long-term impact of CAR on pathophysiological processes. Thus far, data support CAR as an affordable, accessible (over-the-counter) essential oil that may be valuable in countering acute effects of TBI.
No comments:
Post a Comment