http://journal.frontiersin.org/article/10.3389/fnagi.2016.00281/full?
 Alejandro Romero1*,
Alejandro Romero1*,  Eva Ramos1,
Eva Ramos1,  Paloma Patiño2,
Paloma Patiño2,  Maria J. Oset-Gasque3,
Maria J. Oset-Gasque3,  Francisco López-Muñoz4,5 and
Francisco López-Muñoz4,5 and  José Marco-Contelles6*
José Marco-Contelles6*- 1Department of Toxicology and Pharmacology, Faculty of Veterinary Medicine, Complutense University of Madrid, Madrid, Spain
- 2Paediatric Unit, La Paz University Hospital, Madrid, Spain
- 3Department of Biochemistry and Molecular Biology II, Faculty of Pharmacy, Complutense University of Madrid, Ciudad Universitaria, Madrid, Spain
- 4Faculty of Health, Camilo José Cela University, Madrid, Spain
- 5Neuropsychopharmacology Unit, “Hospital 12 de Octubre” Research Institute, Madrid, Spain
- 6Laboratory of Medicinal Chemistry, Institute of General Organic Chemistry (CSIC), Madrid, Spain
Introduction
Stroke is a disease of aging, affecting an increasing number of people worldwide, and the main cause of disability (Flynn et al., 2008; Mathers et al., 2009).
The ischemic cascade begins with the energy failure produced by the
obstruction of a blood vessel that produces a massive and prolonged
release of glutamate (Rothman and Olney, 1986). Physiopathological events associated with brain ischemia are related to oxidative stress process, Ca2+
dyshomeostasis, mitochondrial dysfunction, pro-inflammatory mediators
and/or programmed neuronal cell death. In the ischemic stroke, as the
result of the obstruction of a blood vessel, a critical reduction in the
cerebral blood flow (less than 25%) occurred in brain, and neurons need
a continued supply of oxygen and glucose. Under deprivation of oxygen
and glucose, cell death occurs in two phases: (a) first cell death from
anoxia/hypoxia and energy depletion, followed by; and (b) reperfusion
that increase oxidative stress and free radical formation,
excitotoxicity and nitric oxide (NO) production with ulterior energy
failure and delayed death (Hossmann, 1994; Choi, 1996; Lee et al., 1999; Ito et al., 2003).
No effective therapeutic drugs to treat or prevent brain
damage in ischemic stroke are currently available. Only recombinant
tissue plasminogen activator (r-tPA) is used to open a blood vessel, but
r-tPA has a very narrow therapeutic window of 3.5 h (Zivin et al., 1985).
Preventing brain damage during the ischemic penumbra, despite that it
is a hypoperfused and non-functional tissue, is still a viable tissue
adjacent to the infarcted core. Finally, new therapeutic agents are
needed to recover tissue functionality before cell death, and to be
effective in dealing with several targets, including excitotoxicity and
disturbed calcium ion homeostasis, mitochondrial failure, oxidative and
nitrosative stress, inflammation and apoptosis (Paschen, 2000; Chan, 2001; Iadecola and Alexander, 2001; Lo E. H. et al., 2005; Niizuma et al., 2010). In this Perspective
article, we will focus on melatonin and nitrones, well-known radical
scavenging and antioxidant agents, for the potential therapy of stroke (Hardeland, 2009).
Melatonin
Stroke as a main cause of brain disease arouses great
interest in therapeutic strategies development. The fact that no
effective treatment for stroke has yet been approved to date makes
melatonin a promising molecule for stroke treatment, either alone or in
combination with other agents. A great number of studies had been
developed with melatonin prevention or counteracting stroke damage at
several steps of the ischemic cascade, such as neuroinflammation,
oxidative stress, excitotoxicity and/or apoptosis (Barlow-Walden et al., 1995; Sinha et al., 2001; Rodriguez et al., 2004; Ozacmak et al., 2009; Reiter et al., 2009; Koh, 2012a; Kim and Lee, 2014; Manchester et al., 2015; Zhao et al., 2015; Alluri et al., 2016).
We have recently demonstrated that in rat hippocampal slices subjected
to oxygen-glucose-deprivation (OGD) and glutamate excitotoxicity,
melatonin is able to mediate neuroprotection (Patiño et al., 2016).
Previously, we also demonstrated that melatonin exerts its protective
effect post-ischemia through the nicotinic acetylcholine receptor α7
subunit modulated by an overexpression of heme oxygenase-1 (Parada et al., 2014).
Numerous experimental in vivo studies evidenced
that doses in a range of 5–15 mg/kg of melatonin, mainly administered
intraperitoneally, exert neuroprotective effects in the ischemic cascade
at several critical points (Guerrero et al., 1997; Pei et al., 2003; Pei and Cheung, 2004; Chen H. Y. et al., 2006; Carloni et al., 2008; Signorini et al., 2009; Balduini et al., 2012; Alonso-Alconada et al., 2013; Paredes et al., 2015).
In vivo data confirm the efficacy of this
indoleamine. Melatonin has been related to brain repair by comparing
pinealectomized and non-pinealectomized animals, observing a greater
neurodegeneration in the last group (Manev et al., 1996).
Some studies showed its capacity to counteract oxidative stress
downregulation or scavenging oxygen and nitrogen species and its free
radical detoxification capacity (Guerrero et al., 1997; Pei et al., 2003; Rodriguez et al., 2004; Chen H. Y. et al., 2006; Koh, 2008d).
Other results in stroke models reveal the efficacy of
the antiapoptotic properties of melatonin through several mechanisms
like increasing levels of Bcl-2, blocking caspase cascade or by
preventing mitochondrial depolarization (Sun et al., 2002; Andrabi et al., 2004; Koh, 2008b).
In stroke, elevated extracellular glutamate is critical
in neuronal damage. Herein, melatonin has also demonstrated a
neuroprotective effect in vivo, mitigating Ca2+ influx (Camello-Almaraz et al., 2008) via melatonin receptor (Das et al., 2010) by reducing lipid peroxidation (Kim and Kwon, 1999; Wakatsuki et al., 2001).
Interestingly, melatonin is a free radical scavenger, which inhibits NO
synthesis, a mediator of glutamate and therefore reducing the
excitotoxicity (Chung and Han, 2003).
In animal stroke models, inflammation leads to numerous pathological
events, but melatonin treatment reduces macrophage brain infiltration,
activated microglia prevents IL-1β, TNF-α and GFAP overexpression (Lee et al., 2007; Paredes et al., 2015), which taken together inhibit the inflammatory response.
Nonetheless, blood brain barrier (BBB) integrity is
compromised after cerebrovascular insults, by an increased release of
proinflammatory mediators (COX-2, TNF-α, IL-1β, IL-6), ROS, protein
extravasation and interstitial edema. In animal models, melatonin
significantly reduces BBB dysfunction through several mechanisms, NO,
ROS and RNS levels, preserves tight junction proteins as claudin-5 and
modulates hyperpermeability (Chen H. Y. et al., 2006; Grossetete et al., 2009; Song et al., 2014; Moretti et al., 2015; Alluri et al., 2016). In light of these results melatonin shows a suitable profile to preserve BBB functional integrity.
Among brain cell populations, neural stem cells (NSCs)
have the potential to regenerate new neuronal population. It has been
described that after a melatonin treatment, neurogenesis is induced
through melatonin receptor MT2 (Chern et al., 2012).
Despite molecular neuroprotective mechanisms are not well defined,
melatonin has demonstrated to enhance neurogenic cells of the ischemic
brain, in striatum neurons and the hippocampal region (Kilic et al., 2008; Ayao et al., 2010; Lee et al., 2014).
Furthermore, mesenchymal stem cells (MSCs) are used in implantations
after the ischemic insult, but unfortunately this procedure involves the
difficulty that approximately the 80% of the grafted cells do not
survive (Roh et al., 2008). Melatonin pre-administration achieves a higher percentage of MSCs survival, also through a receptor-mediated mechanism (Tang et al., 2014).
As far as signal transduction pathways are involved in
stroke, melatonin has emerged as a versatile neuroprotective regulator.
Melatonin neuroprotective effects are achieved through receptor-mediated
mechanisms (MT1, MT2 and MT3; Reiter et al., 2007; Tan et al., 2007; Slominski et al., 2012; Lacoste et al., 2015). Activation of MT1 melatonin receptor leads to the stimulation of a large variety of G proteins (Brydon et al., 1999), upregulation of MT2 promoted neurogenesis and preservation of BBB integrity (Chern et al., 2012) and stimulation of MT3 may contribute to the antioxidant potential of melatonin (Tan et al., 2007).
In addition, melatonin is highly effective in preventing Ca2+ dyshomeostasis during ischemic brain injury (Camello-Almaraz et al., 2008; Koh, 2012b).
The antiapoptotic effect of melatonin in brain ischemia models is
related to its actions at the mitochondria level, preventing the
injury-induced reduction of pBad levels and the mitochondrial
depolarization inhibiting the mitochondrial permeability transition pore
(mPTP; Andrabi et al., 2004; Kilic et al., 2004b; Koh, 2008b).
Cell proliferation, differentiation, survival and apoptosis are
regulated by the PI3K/Akt signaling pathway and activation of iNOS
signaling is associated with PI3K/Akt inhibition. It has been reported
that melatonin upregulates this pathway, and decreases iNOS levels (Kilic et al., 2005; Koh, 2008c,d).
Matrix metalloproteinases (MMPs) are a family of calcium-dependent
zinc-binding proteolytic enzymes that degrade the extracellular matrix
(ECM) components of the basement membrane (Montaner et al., 2001). Administration of exogenous melatonin attenuated postischemic MMP-9 activation reducing brain damage in stroke models (Hung et al., 2008).
The mitogen-activated protein (MAP) kinase/extracellular-regulated
kinase (ERK) 1/2 signaling pathway regulates cell differentiation,
growth, survival and apoptosis (Pearson et al., 2001).
It has been reported that melatonin plays neuroprotection through Akt
and ERK1/2 phosphorylation, and activates MEK/ERK/p90RSK/Bad cascade
signaling (Kilic et al., 2005; Koh, 2008a).
After hypoxic/ischemic brain injury, endogenous vasoconstrictor
endothelin-1 (ET-1) levels are elevated leading to exacerbated brain
injury, but when melatonin is administered in mice stroke models a
beneficial neuroprotective effect was observed inhibiting ET-1 (Kilic et al., 2004a; Lo A. C. et al., 2005). Phosphorylation/dephosphorylation processes are the major form of cellular signaling (Gong and Iqbal, 2008), and their deregulation turns in severe pathologies including neurodegeneration (Sontag et al., 2004), cancer (Ruediger et al., 2001), cardiovascular (Ling et al., 2012) and metabolic disorders (Mandavia and Sowers, 2012). The phosphoprotein phosphatase 2A (PP2A) is the principal member of the family of Ser/Thr phosphatases (Liu et al., 2005),
which removes phosphate from serine and threonine residues. Several
compounds may activate or protect PP2A enzyme activity and have
neuroprotective actions in in vivo and in vitro models of brain damage (Shah et al., 2015).
In this context, melatonin exerts an upregulation of PP2A enzyme
activity, which implies that the PP2A malfunction observed in
excitotoxic environments could be mitigated by the administration of
melatonin (Koh, 2012a). The protective effect of Silent information regulator 1 (SIRT1) on the brain has been well demonstrated (Yang et al., 2013).
Melatonin preserves SIRT1 expression, activates SIRT1 signaling in
neuronal cells after hypoxia-ischemia attenuating mitochondrial
oxidative damage (Carloni et al., 2014; Yang et al., 2015).
Finally, melatonin has been combined with other drugs, such as t-PA (Chen T. Y. et al., 2006), topiramate (Ozyener et al., 2012), nimodipine and other Ca2+ antagonists (Gelmers et al., 1988; Toklu et al., 2009), meloxicam (Gupta et al., 2002) for stroke therapy, giving promising results.
Nitrones
Based on the understanding of the biochemical processes
involved in the formation and development of a stroke, number of
products have been developed targeting the different ischemic and
reperfusion events. Despite the promising initial results,
neuroprotection drugs for stroke have failed in advanced clinical
phases, and consequently, no neuroprotective agent has been approved by
the FDA for stroke therapy. However, neuroprotection is still a choice,
and oxidative stress, a suitable biological target. In this context,
antioxidants such as N-t-butylphenylnitrone (PBN; Figure 1; Novelli et al., 1986) and NXY-059 (Figure 1; Dehouck et al., 2002), have attracted the interest of a number of laboratories, resulting in therapeutic candidates for cancer (Inoue et al., 2007; Floyd et al., 2008, 2010, 2011; Costa et al., 2015), neurodegenerative disorders (Floyd et al., 2000), hearing loss (Floyd et al., 2008) and stroke (Doggrell, 2006; Floyd et al., 2008). NXY-059 (Figure 1) (Kuroda et al., 1999)
is a well-known free radical scavenger with good neuroprotective
profile in rat models of transient and permanent focal ischemia, and
stroke model in rodents, which has been launched several times in
different program in advanced clinical studies, although with limited
success (Macleod et al., 2008). In fact and in addition, tert-butylnitrones, such as NXY-059, are known to afford t-butylhydroxylamines
as powerful radical scavengers, after hydrolysis, that further could be
oxidized to 2-methyl-2-nitrosopropane which then may synthesize NO
radical, the source and origin of the neuroprotection, as it has been
already reported for NO donors (Godínez-Rubí et al., 2013).
Recently reported new developments have highlighted the not previously
described and powerful neuroprotective effect shown by new PBN
derivatives bearing N-aryl substituents on human neuroblastoma cells, under induced in vitro experimental oxidative stress (Matias et al., 2016).
FIGURE 1
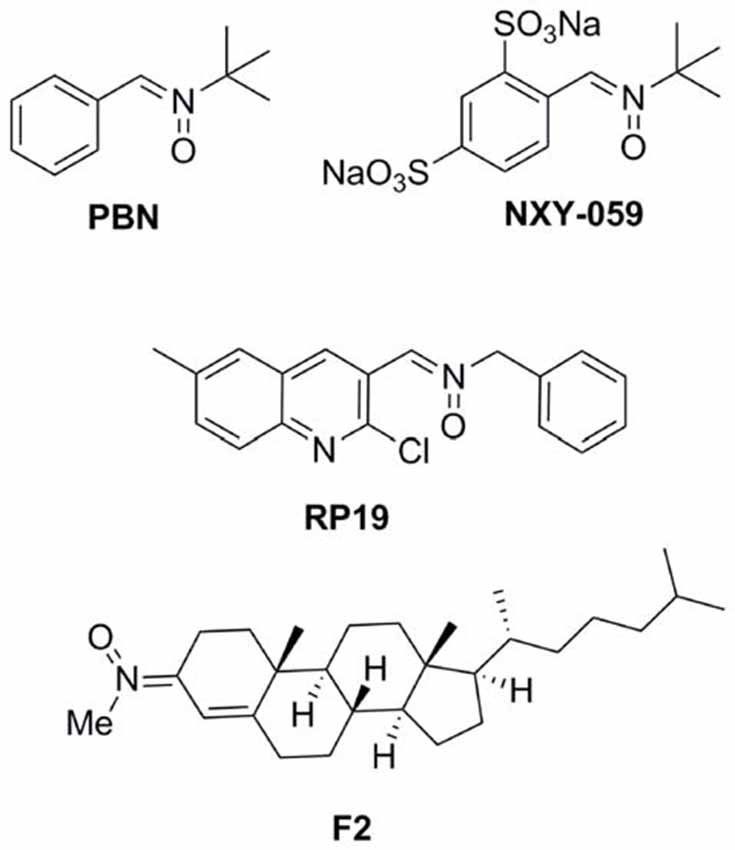 Figure 1. Structures of nitrones N-t-butylphenylnitrone
(PBN), NXY-059, and the nitrones RP19 and F2, assesed in our
laboratories.
Figure 1. Structures of nitrones N-t-butylphenylnitrone
(PBN), NXY-059, and the nitrones RP19 and F2, assesed in our
laboratories.
Starting in 2008, the group led
by Marco-Contelles (CSIC, Madrid, Spain) has designed, synthesized and
developed a number of nitrones for the potential treatment of stroke,
the most interesting compounds being RP19 (Figure 1; Chioua et al., 2012), and F2 (Ayuso et al., 2015; Figure 1),
in collaboration with Dr. Dimitra J. Hadjipavlou-Litina (Aristotle
University of Thessaloniki, Greece) and Dr. Alcázar (Hospital Ramón y
Cajal, Madrid, Spain).
As radical-trapping agents, nitrones are expected to
delay or prevent oxidation of easily oxidizable substrates, therefore
being considered antioxidants. In vitro radical trapping and
antioxidant activity were studied for nitrones RP19 and F2, and PBN as
reference compound, using the DPPH quenching, •OH and O∙−2
scavenging activities were low in general, with moderate values for
RP19 (42.3% and 23%). Scavenging of •OH, as one of the most toxic
radicals generated during ischemic stress, was also determined, showing
that higher trapping activities were achieved than reference compound
PBN.
TABLE 1A
 Table 1A. In vitro antioxidant activity (A) and neuroprotection in neuronal cultures and in vivo model of ischemia (B).
Table 1A. In vitro antioxidant activity (A) and neuroprotection in neuronal cultures and in vivo model of ischemia (B).
Testing in neuronal cultures and in in vivo
experiments were next evaluated. Thus, the neuroprotective effect of
nitrones RP19, F2, as well as PBN and NXY-059 as standards were studied
in primary neuronal cultures, which were subjected to OGD as an in vitro
model of ischemia. Cell viability was evaluated by quantification of
living, metabolically active cells, as determined by the MTT assay.
Neuroprotection is expressed as the percentage to reach the control
value (100%), from the untreated ischemic neurons value (0%) (Table 1B).
As shown, all the nitrones tested afforded values in all cases higher
than the one determined for PBN (13.4 ± 1.9% at 5 mM) and NXY-059 (56.8 ±
2.5% at 250 μM), being remarkable for those observed for nitrones RP19
(87.5 ± 3.2% at 50 μM) and F2 (80.7 ± 2.7% at 5 μM). Next, transient
global brain ischemia was performed on adult rats by the standard
four-vessel occlusion model, in which carotid arteries are occluded
during 15 min and 24 h after the irreversible occlusion of both
vertebral arteries by electrocoagulation (Pulsinelli and Brierley, 1979; García-Bonilla et al., 2007).
Ischemic animals were treated with RP19 and F2; NXY-059 diluted in 10%
ethanol in saline as a vehicle by intraperitoneal injection when carotid
arteries were un-clamped for reperfusion. Animals were studied after 5
days of reperfusion (R5d) after killing by transcardiac perfusion
performed under deep anesthesia. Treatments were blindly and randomly
performed and body temperature of 37°C was maintained (Table 1B). Cell death and apoptosis were assessed in the hippocampal cornu ammonis
1 (CA1) region and cerebral cortex (C). Nitrones RP19 and F2 showed
higher inhibition of cell death than for NXY-059. In particular, best
results were obtained with F2 at 0.1 mg/Kg concentration, and RP19 at
0.5 mg/Kg concentration. Apoptosis reduction by F2 (35% in hippocampal
CA1, 91% in cortex, at 0.1 mg/kg concentration) and RP19 (38% in
hippocampal CA1, 79% in cortex, at 0.5 mg/kg concentration) showed the
best results, both higher than the values observed for NXY-059 (21% in
hippocampal CA1, 55% in cortex), at the same concentration.
No comments:
Post a Comment