Your doctors and hospital need to ensure followup research occurs
Targeting Parthanatos in Ischemic Stroke
 Raymond C. Koehler
Raymond C. Koehler Valina L. Dawson
Valina L. Dawson Ted M. Dawson
Ted M. Dawson- 1Department of Anesthesiology and Critical Care Medicine, The Johns Hopkins University, Baltimore, MD, United States
- 2Neuroregeneration and Stem Cell Programs, The Institute of Cell Engineering, The Johns Hopkins University, Baltimore, MD, United States
- 3Department of Neurology, The Johns Hopkins University, Baltimore, MD, United States
- 4Department of Neuroscience, The Johns Hopkins University, Baltimore, MD, United States
- 5Department of Physiology, The Johns Hopkins University, Baltimore, MD, United States
- 6Department of Pharmacology and Molecular Sciences, The Johns Hopkins University, Baltimore, MD, United States
Parthanatos is a cell death signaling pathway in which excessive oxidative damage to DNA leads to over-activation of poly(ADP-ribose) polymerase (PARP). PARP then generates the formation of large poly(ADP-ribose) polymers that induce the release of apoptosis-inducing factor from the outer mitochondrial membrane. In the cytosol, apoptosis-inducing factor forms a complex with macrophage migration inhibitory factor that translocates into the nucleus where it degrades DNA and produces cell death. In a review of the literature, we identified 24 publications from 13 laboratories that support a role for parthanatos in young male mice and rats subjected to transient and permanent middle cerebral artery occlusion (MCAO). Investigators base their conclusions on the use of nine different PARP inhibitors (19 studies) or PARP1-null mice (7 studies). Several studies indicate a therapeutic window of 4–6 h after MCAO. In young female rats, two studies using two different PARP inhibitors from two labs support a role for parthanatos, whereas two studies from one lab do not support a role in young female PARP1-null mice. In addition to parthanatos, a body of literature indicates that PARP inhibitors can reduce neuroinflammation by interfering with NF-κB transcription, suppressing matrix metaloproteinase-9 release, and limiting blood-brain barrier damage and hemorrhagic transformation. Overall, most of the literature strongly supports the scientific premise that a PARP inhibitor is neuroprotective, even when most did not report behavior outcomes or address the issue of randomization and treatment concealment. Several third-generation PARP inhibitors entered clinical oncology trials without major adverse effects and could be repurposed for stroke. Evaluation in aged animals or animals with comorbidities will be important before moving into clinical stroke trials.
Introduction
With the recent success of endovascular thrombectomy in establishing recanalization and, in many cases, reperfusion in stroke patients with large vessel occlusions, neurologic outcome has been improved, especially for those who have sufficient collateral blood flow to slow the transition from penumbra to irreversible injury (1–7). However, a significant portion of those with successful reperfusion may still die or be left with significant neurologic deficits. Thus, interest has been renewed in finding neuroprotective agents that can be used as an adjunct with thrombectomy and clot lysis (8–10).
One potential target that has been investigated in the context of cerebral ischemia over the past three decades is poly(ADP-ribose) polymerase (PARP). This family of enzymes is involved in post-translational protein modification by ADP-ribosylation. PARP1, in particular, is well-known for its role in the DNA repair process. Indeed, prolonged inhibition of PARP1 is cytotoxic and has been used as a strategy in oncology to induce cell death in rapidly proliferating cancer cells. However, when numerous DNA strands are damaged and PARP activity reaches high levels, this elevated activity can induce programmed cell death. This PARP-dependent form of programmed cell death has been termed parthanatos. A number of PARP inhibitors have been developed that block parthanatos, including that which occurs in neurons in response to excitotoxic stimuli and oxygen-glucose deprivation (OGD). A body of literature indicates that, in addition to arresting cell death, PARP inhibitors can reduce the NFκB-mediated pro-inflammatory response of microglia, suppress matrix metalloproteinase (MMP-9) release, and limit blood-brain barrier (BBB) damage and hemorrhagic transformation. Thus, PARP inhibitors can act to directly protect neurons and to limit secondary injury from inflammatory processes. These multi-targeted actions of PARP inhibitors make them strong candidates for prolonging neuronal viability and ameliorating reperfusion injury. In this review, we will consider the mechanisms by which PARP activity contributes to cell death and neuroinflammation and then examine the evidence that PARP inhibitors can protect the brain from ischemic stroke.
Mechanisms of PARP-Dependent Neuronal Cell Death (Parthanatos)
Damage to DNA by oxidants such as peroxynitrite rapidly activates PARP (11, 12). Peroxynitrite formation is increased by the excess production of nitric oxide (NO) and superoxide anion (13–15). In the context of cerebral ischemia, reduced ATP production results in neuronal depolarization, release of synaptic glutamate, and decreased reuptake by glutamate transporters (16, 17). Excess extracellular glutamate activates N-methyl-D-aspartate (NMDA) and other glutamate receptors, leading to an excitotoxic process that involves increased intracellular Ca2+. A key aspect of the excitotoxic process is the association of the NR2B subunit to post-synaptic density-95 scaffolding protein and stimulation of NO synthase (18). NO is highly diffusible and can target other neurons and cell types. Major sources of superoxide during and after ischemia are the uncoupling of the mitochondrial electron transport chain (19) and the activation of NADPH oxidases (20, 21). In the context of reperfusion, the increased reoxygenation supports excess superoxide production (22). Although regeneration of ATP can reduce extrasynaptic glutamate detected by microdialysis, it is thought that increased glutamate levels in the extracellular synaptic cleft can persist during reperfusion (23, 24). In addition, altered phosphorylation of NMDA receptor subunits and of neuronal nitric oxide synthase (nNOS) can sustain the activity of these molecules through their persistent association with post-synaptic density proteins during the reperfusion period (25). Thus, peroxynitrite formation can be augmented during the reperfusion period and lead to further DNA damage and activation of PARP. This scenario provides a basis for the therapeutic potential of PARP inhibitors administered during reperfusion produced by thrombectomy and tissue plasminogen activator administration.
In 1994, the Dawson laboratory reported that exposing cultured mouse neurons to NMDA or high levels of NO activates PARP and that inhibition of PARP protects the neurons from cell death (26). Moreover, PARP1−/− and nNOS−/− murine neurons were shown to be resistant to cell death evoked by NMDA (27, 28). These findings have more recently been replicated in human neuronal cultures derived from embryonic stem cells and inducible pluripotent stem cells, as these cultures were protected from NMDA and OGD by inhibition of PARP or nNOS (29). Importantly, protective effects were seen in both male and female human neurons with the new generation of PARP inhibitors developed for use in oncology: olaparib, veliparib, rucaparib, and talazoparib. PARP's instigation of cell death involves release of apoptosis-inducing factor (AIF) from a mobile pool in the outer mitochondrial membrane (30–32). This release requires the formation of large PAR polymers by PARP and the binding of PAR to a PAR-binding site motif on AIF (33). Degradation of the PAR polymer by PAR glycohydrolase (PARG) decreased the release of AIF by PAR polymers (34) and by NMDA (35). Once AIF is released from the mitochondria, it can bind to macrophage migration inhibitory factor (MIF) (36). The binding of AIF to MIF results in uptake of the complex into the nucleus where the complex binds to DNA. The binding enables MIF to act as an endonuclease that degrades genomic DNA into 20–50 kb DNA fragments, and that leads to cell death. This signaling pathway is summarized in Figure 1 and is reviewed in detail elsewhere (37).
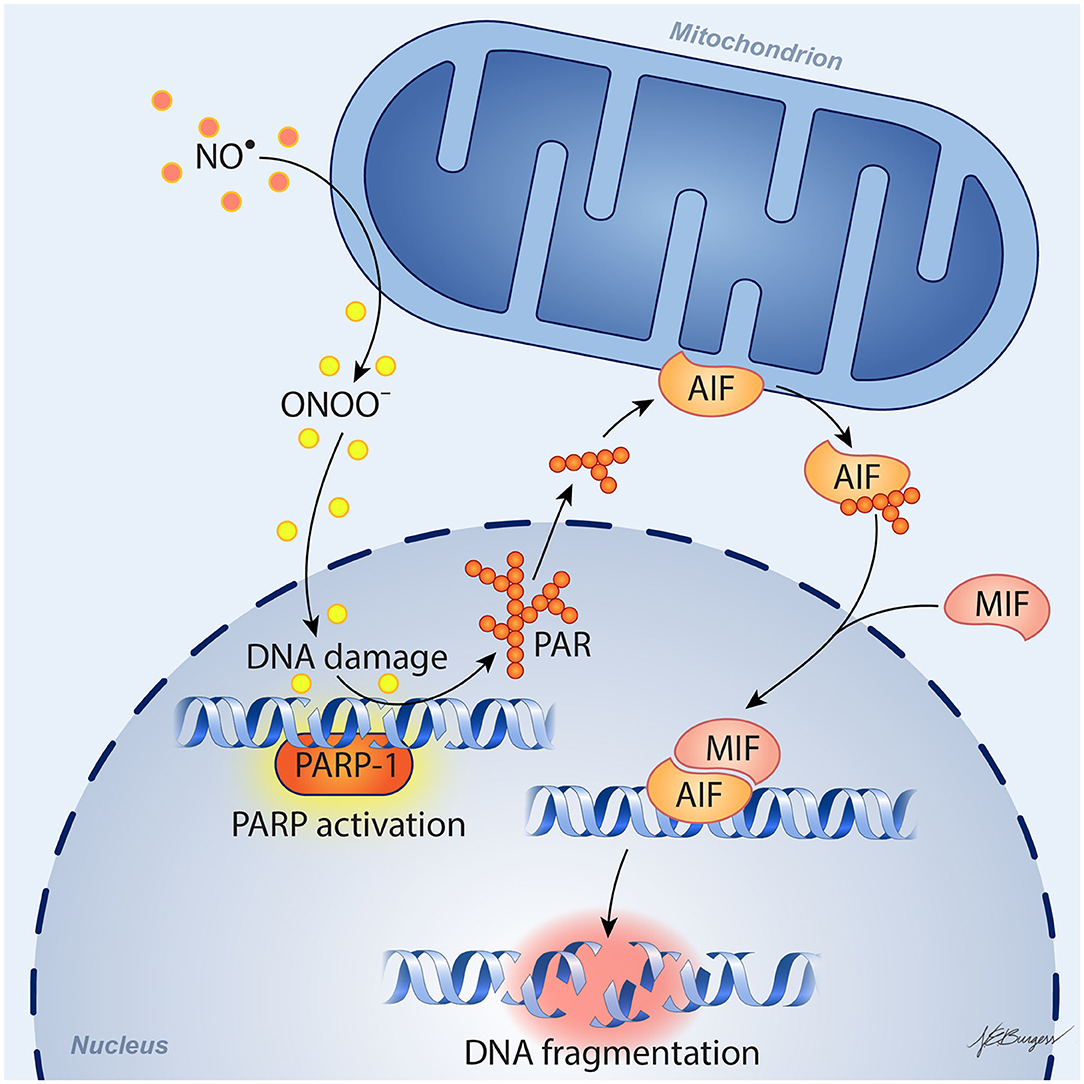
Figure 1. Schematic diagram of parthanatos signaling. Activation of NMDA receptors stimulates neuronal nitric oxide (NO) synthase (nNOS). Ischemia and reperfusion lead to excess superoxide production in mitochondria and from NADPH oxidase. The abundant NO and superoxide spontaneously generate peroxynitrite (ONOO−). This strong prooxidant, along with other reactive oxygen species (ROS), damages DNA strands and thereby causes activation of poly(ADP-ribose) polymerase (PARP). When the DNA damage is great, the consequent over-activation of PARP leads to abundant PAR polymer formation in the nucleus; some of the poly(ADP) ribosylated carrier proteins exit the nucleus and cause the release of apoptosis-inducing factor (AIF) from a pool on the outer mitochondrial membrane. Once in the cytosol, AIF can bind to macrophage migration inhibitory factor (MIF). Together, they enter the nucleus and produce large-scale DNA degradation and cell death.
Prolonged ischemia results in classical necrosis, whereas reperfusion can result in sustained restitution of cell function if the duration and severity of ischemia are not prolonged or intense. With reperfusion after an intermediate ischemic duration and/or intermediate ischemic severity, some neurons can generate sufficient ATP to restore membrane potential. Often, these neurons then undergo various forms of delayed programmed cell death. Delayed apoptosis may occur in ischemic border regions where ischemia is less severe and not as prolonged. However, with durations of middle cerebral artery (MCA) occlusion (MCAO) lasting >30 min, programmed necrosis predominates. The two most studied forms of programmed necrosis in stroke are parthanatos and RIP1-dependent necroptosis. The latter involves formation of a necroptosome consisting of phosphorylated RIP-1 and phosphorylated RIP-3 anchored by MLKL. Inhibition of necroptosome formation by intraventricular injection of necrostatin-1 decreases infarct volume by one-third (38), but clinical translation of this work is limited by the poor BBB penetration of this inhibitor class. In contrast, several different PARP inhibitors have proven effective following systemic administration, yielding a reduction in infarct volume that often exceeds 50%, as reviewed below.
In addition to the parthanatos mechanism of neurodegeneration described above, the product of PARP activation, PAR, is emerging as an important contributor in the separation and aggregation of amyloid proteins in the degenerative process of Parkinson's disease, Alzheimer's disease and amyotrophic lateral sclerosis (39). Future studies investigating the role of PAR dependent liquid-liquid phase separation and aggregation of proteins are needed to determine whether this plays a role in stroke pathogenesis.
Mechanisms of PARP-Dependent Neuroinflammation
In addition to acting directly on neurons to block parthanatos, PARP inhibitors can exert anti-inflammatory effects and protect endothelial cells. For example, PARP inhibitors have been reported to exert anti-inflammatory effects in a wide range of systemic models of inflammation (endotoxemia, peritonitis, colitis, streptozotocin-induced diabetes) (40) and to protect endothelial-dependent responses from oxidant stress associated with aging (41), streptozotocin-induced diabetes (42), hypochlorite (43), and H2O2 (44). Exposing macrophages to oxidants stimulates their PARP activity (45). Exposing cultured BV2 microglial cells to lipopolysaccharide induces NF-κB, inducible nitric oxide synthase (iNOS), nitrite formation, reactive oxygen species formation, and tumor necrosis factor-α (TNFα), all of which are attenuated by the PARP inhibitor PJ34 (46). With global cerebral ischemia, PJ34 pre-treatment attenuated loss of tight junctions, Evan's Blue extravasation, cerebral edema, and neutrophil infiltration (47). Microglia-mediated loss of tight junctions is absent in co-cultures of PARP1-null microglia and endothelial cells (48). In cultured human brain microvascular endothelial cells, PARP inhibitors reduce AIF nuclear translocation and cell death after repeated bouts of hypoxia and reoxygenation (49). In microglial cultures exposed to the alarmin S100B, induction of interleukin-1β (IL-1β) and iNOS was blunted by the PARP inhibitor veliparib, and injection of S100B directly into striatum produced an accumulation of Iba1-positive microglia that was attenuated by veliparib (50). Glutamate activation of microglia is also PARP-dependent (51).
It is known that enzymatic activity of PARP1 promotes NF-κB–driven transcription in microglia (52). The underlying mechanism has been investigated by Kauppinen et al. In microglia, PARP1 activity facilitates NF-κB binding to DNA, and PARP1 gene deletion or inhibition suppresses TNFα-induced release of MMP-9 by microglia and toxicity to co-cultured neurons (53). As illustrated in Figure 2, TNFα activation of PARP1 involves Ca2+ influx, stimulation of phosphatidylcholine-specific phospholipase C, and activation of the MEK1/2-ERK1/2 pathway (54), leading to phosphorylation of PARP1 by ERK2 (55). This mechanism does not require DNA damage, which can independently activate PARP via stimulation of ERK1/2. One way PARP activity might enable NF-κB binding to DNA is by competing with sirtuin 1 for NAD+ substrate. When sirtuin 1 deacetylates the p65 subunit of NF-κB, it is not able to bind to DNA. With excessive activity of PARP sufficient to decrease NAD+, NF-κB binding to DNA is increased. This effect is reversed by preventing the decrease in NAD+ and is replicated by inhibition of sirtuin 1 (56). Thus, enzymatic activity of PARP1 plays a key role in the activation of NF-κB binding to DNA.
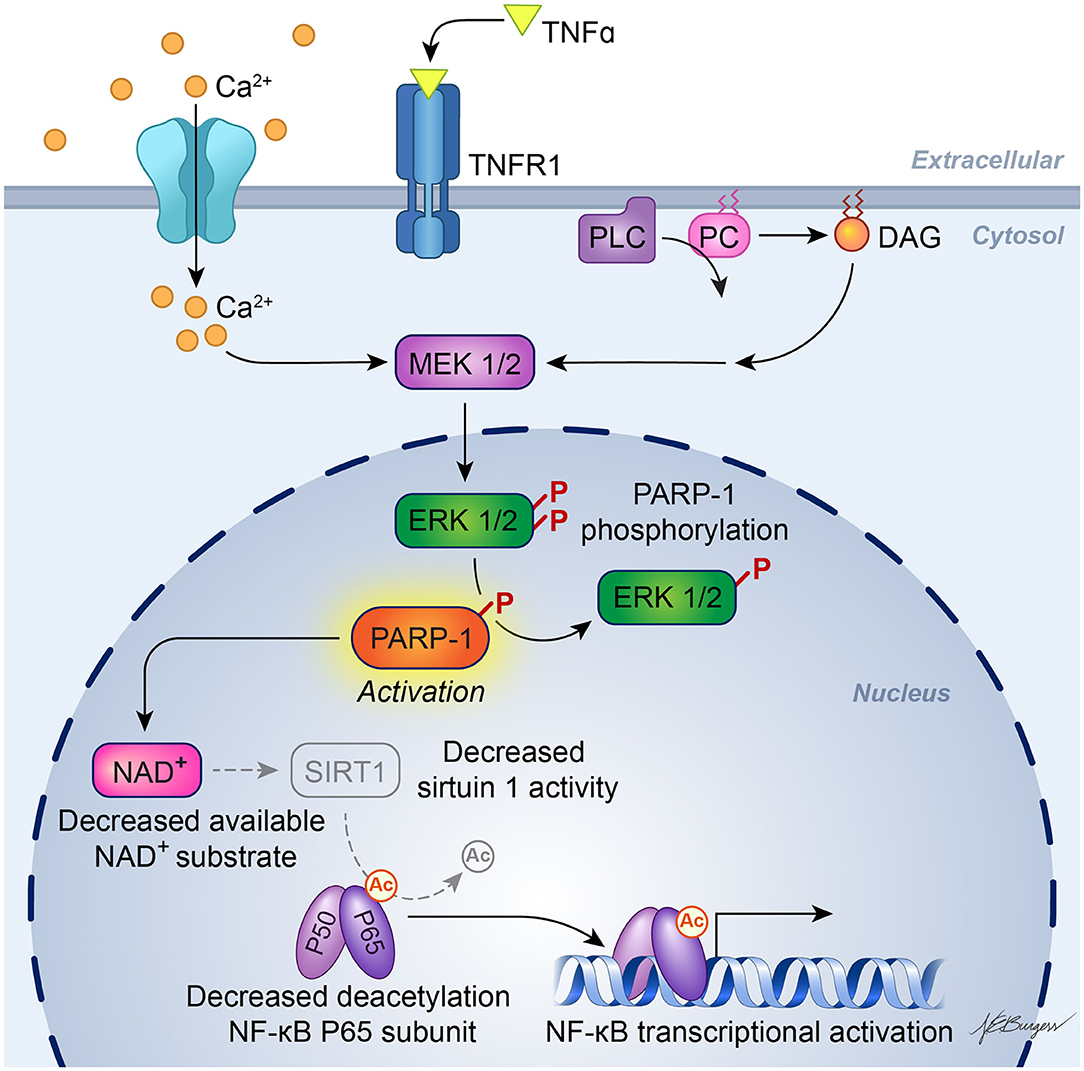
Figure 2. Schematic diagram of PARP activation in inflammatory signaling. Experiments in cultured microglia and astrocytes indicate that binding of TNFα to its receptor, TNFR1, leads to calcium influx and activation of phosphatidylcholine (PC)-specific phospholipase C (PC-PLC), resulting in formation of diacylglycerol (DAG) and downstream action of the MEK1/2-ERK1/2 pathway. ERK2 phosphorylation of PARP-1 increases PARP-1 activity, which then facilitates activation of NF-κB transcription. One possible mechanism whereby PARP-1 facilitates NF-κB transcription is by depleting NAD+, which is also used as a substrate by sirtuin-1. Sirtuin-1 normally keeps the P65 subunit of NF-κB deacetylated, but a decrease in sirtuin-1 activity will result in acetylated P65, which enables NF-κB transcription. [Adapted, with permission, from Vuong et al. (54) and Kaupinnen et al. (56)].
In vivo, several pieces of evidence are consistent with PARP inhibitors acting directly to modify the delayed neuroinflammatory response in models of traumatic brain injury (TBI) or ischemia. Rats treated with the PARP inhibitor INO-1001 starting 1 day after TBI exhibited reduced activation of microglia (57), and mice treated with PJ34 starting 1 day after TBI had less neuronal loss in cortex and thalamus and improved sensorimotor function at 3 weeks of recovery (46). Likewise, veliparib decreased expression of pro-inflammatory cytokines and Iba-1 in rats and Iba-1 expression in pigs, even when administration was delayed until 24 h after TBI (58). In a model of global ischemia, administration of PJ34 at 8 h of reperfusion reduced microglial activation while increasing neuronal survival at 7 days (59), and a further delay in treatment until 2 days still resulted in suppressed microglial activation and astrogliosis and increased neuronal density and bromodeoxyuridine staining 8 weeks later (60). With regard to transient MCAO, pre-treatment with PARP inhibitor 3-aminobenzamide (3-AB) decreased MMP-9 activity, neutrophil infiltration, and infarct volume in rats (61), and PJ34 reduced the induction of iNOS and ICAM-1 and decreased infarct volume at 72 h in mice (62). Minocycline is an inhibitor of PARP1 at nanomolar concentrations (63), and this inhibition may be one mechanism by which minocycline reduces post-ischemic neuroinflammation. Finally, a clinical study showed that treating blood samples from stroke patients ex vivo with a PARP inhibitor increased the fraction of T-regulatory cells, which are thought to play a role in the brain repair stage after stroke (64).
Therefore, PARP inhibitors are likely to be multipotent in stroke by (1) blocking programmed neuronal necrosis in a large portion of neurons, (2) attenuating the early pro-inflammation response that is thought to be accelerated by reperfusion, and (3) protecting the endothelium and limiting hemorrhagic transformation that is thought to be augmented during aging. This broad spectrum action may be superior to that of drugs such as nerinetide (Tat-NR2B9c), which primarily targets excitotoxicity (18, 65, 66) and failed in the recent Phase III ESCAPE-NA1 trial (67) of ischemic stroke reperfusion therapy.
No comments:
Post a Comment