Oh God, what stupidity. Changing wording
from neuroprotection to Brain Cytoprotection. It still doesn't sound
important enough to immediately fix. Neuronal cascade of death implies immediate interventions are needed. And you don't mention trying to stop the 5 causes of the neuronal cascade of death in the first days. I see zero hope in this with NO stroke leadership and NO stroke strategy.
Pharmacological brain cytoprotection in acute ischaemic stroke — renewed hope in the reperfusion era
Nature Reviews Neurology (2022)
Abstract
For over 40 years, attempts to develop treatments that protect neurons and other brain cells against the cellular and biochemical consequences of cerebral ischaemia in acute ischaemic stroke (AIS) have been unsuccessful. However, the advent of intravenous thrombolysis and endovascular thrombectomy has taken us into a new era of treatment for AIS in which highly effective reperfusion therapy is widely available. In this context, cytoprotective treatments should be revisited as adjunctive treatment to reperfusion therapy. Renewed efforts should focus on developing new drugs that target multiple aspects of the ischaemic cascade, and previously developed drugs should be reconsidered if they produced robust cytoprotective effects in preclinical models and their safety profiles were reasonable in previous clinical trials. Several development pathways for cytoprotection as an adjunct to reperfusion can be envisioned. In this Review, we outline the targets for cytoprotective therapy and discuss considerations for future drug development, highlighting the recent ESCAPE-NA1 trial of nerinetide, which produced the most promising results to date. We review new types of clinical trial to evaluate whether cytoprotective drugs can slow infarct growth prior to reperfusion and/or ameliorate the consequences of reperfusion, such as haemorrhagic transformation. We also highlight how advanced brain imaging can help to identify patients with salvageable ischaemic tissue who are likely to benefit from cytoprotective therapy.
Key points
-
Highly successful reperfusion therapy with intravenous thrombolysis and endovascular thrombectomy is now widely available for the treatment of acute ischaemic stroke, making cytoprotective therapy a viable additional treatment approach.
-
Previous attempts to develop cytoprotective therapy have been unsuccessful, but this approach should now be reconsidered as an adjunctive therapy to thrombolysis and thrombectomy.
-
New cytoprotective drugs should be developed to target multiple aspects of the ischaemic cascade, and previously developed drugs should be reconsidered.
-
Trials should be conducted to evaluate the effects of cytoprotective drugs when administered before or after reperfusion therapy or both.
-
Advanced brain imaging should be used to select patients who are most likely to benefit from cytoprotective treatment for enrolment in new trials.
Introduction
The treatment of acute ischaemic stroke (AIS) has been revolutionized by the development of therapies — such as thrombolysis and endovascular thrombectomy — that open occluded cerebral vessels and restore perfusion to the ischaemic brain. Reperfusion salvages ischaemic brain tissue by restoring the supply of oxygen and glucose and arresting the ischaemic cascade initiated by a lack of these vital nutrients. An additional approach in the treatment of AIS is to directly target the ischaemic cascade in the ischaemic region1. Traditionally, this approach has been referred to as neuroprotection, but the term cytoprotection is more appropriate because all cells of the neurovascular unit — including neurons, astrocytes, microglia, pericytes and the endothelia — in the ischaemic region are at risk of damage2,3. Nevertheless, among these cells, neurons are likely to be the most vulnerable and their death is probably the most important contributor to clinical deficits in AIS2.
Cytoprotective therapies for AIS have remained elusive — a long history of failures and disappointments spans more than 40 years. Many pharmaceuticals that target different components of the ischaemic cascade have produced promising results in animal models of stroke with varying degrees of methodological rigour, but none has had clear, reproducible efficacy on primary clinical end points in clinical trials. Reasons for the lack of translational success have been thoroughly discussed for more than 20 years, and include broad deficiencies in preclinical modelling and clinical trial design4 (Box 1). Most clinical trials of neuroprotective approaches have been designed to evaluate one agent in comparison with placebo, and most enrolled patients have not received concomitant thrombolysis and were unlikely to have had substantial reperfusion before or after the neuroprotective drug was administered5. Given that eventual reperfusion is probably a necessity for neuroprotection to be clinically beneficial, the use of neuroprotective agents in isolation is likely to be a major reason for their failure to date.
In this context, the development of cytoprotective drugs should be reconsidered in the current era of highly successful reperfusion, as use of such drugs as an adjunct to thrombolysis, thrombectomy or both considerably increases the likelihood of success. Indeed, use of such an approach with the cytoprotective drug nerinetide in combination with thrombectomy was evaluated in a recent clinical trial6 and seems to have produced therapeutic benefits in some patients, providing new hope. In this Review, we consider how cytoprotection for AIS can now be developed in the era of acute stroke therapy, taking into account recent developments in imaging and advances in health-care delivery services. We discuss the trial of nerinetide in detail and consider the lessons learned and the next steps towards finally achieving approval of a cytoprotective drug for the clinical management of AIS.
Box 1 Potential preclinical and clinical explanations for failure of neuroprotective drugs
Preclinical
-
Lack of randomized and blinded outcome assessment.
-
Lack of allocation concealment so that investigators were aware of the treatment assignment.
-
Lack of monitoring of physiological measurements, such as blood pressure and temperature.
-
Inappropriate estimation of required sample size.
-
Treatment effects not assessed in animals with comorbidities and old age.
-
Results from all included animals not reported in the study.
-
Lack of dose–response and therapeutic time window studies.
-
Lack of reproducibility between models and laboratories.
-
Incomplete understanding of the pharmacological profile of the drug being tested.
Clinical
-
Drugs were tested in patients who were unlikely to benefit.
-
Studies were performed in an insufficient number of patients to detect a treatment effect.
-
Studies included too many patients with mild or severe stroke, in whom a clinical benefit is difficult to detect.
-
Drug induced serious adverse events that did not allow adequate serum concentrations to be achieved.
-
Trials included a substantial number of patients with lacunar stroke with white matter infarction and no evidence suggests that the study drug affects white matter injury.
-
Patients were assigned to treatment too late after stroke onset to allow substantial ischaemic tissue salvage
-
Lack of advanced imaging in trials of treatment in later time windows to confirm whether the infarct size precluded beneficial treatment.
Therapeutic targets
The ischaemic cascade is a multifaceted and complex pathophysiological process with a multitude of interconnected pathways that evolve over time and involve different cell types that are activated in concert or sequentially by marked reductions in oxygen and glucose supply7 (Fig. 1). Among the most salient components of this process are glutamate-mediated excitotoxicity, calcium signalling, oxidative stress and inflammation that can originate from peripheral immune cells that migrate to the brain and/or from the neurovascular unit at the level of the microglia and endothelium. Neuronal injury and trafficking of peripheral immune cells into the CNS amplifies microglial activation, which results in further neuronal damage. In parallel, neuroprotective responses are initiated, in which astrocytes and microglia respond to so-called ‘help me’ signals from injured neurons by releasing trophic factors and extracellular vesicles and by transferring mitochondria to neurons. The complexity of the ischaemic cascade cannot be overstated. Cell-to-cell signalling among the components of the neurovascular unit either amplify injury or promote protection of neurons, and some components of the intercellular signalling can change from being detrimental to being beneficial or vice versa over the course of hours or days after stroke onset. In addition, extracellular signalling can disrupt the blood–brain barrier (BBB), which can lead to haemorrhagic transformation as a consequence of reperfusion injury (see below). (Generic crap listed. DAMN IT ALL will you solve stroke instead of beating around the bushes? Hell, I'm stroke disabled and I can tell you know fucking nothing about how to solve stroke.)
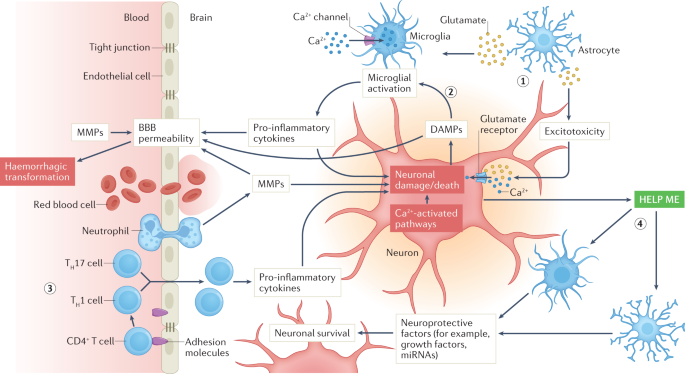
Only selected elements are shown, and the signalling evolves over time after the onset of ischaemia. Oxygen and glucose deprivation leads to release of glutamate from astrocytes (1), which activates excitotoxic signalling within neurons, leading to neuronal injury and death, and activates microglia. Neuronal injury leads to the release of damage-associated molecular patterns (DAMPs), which also activate microglia (2), which consequently release pro-inflammatory cytokines that lead to further damage of neurons and promote endothelial activation and blood–brain barrier (BBB) opening. DAMPS also directly promote BBB breakdown. Leukocyte trafficking and migration to the ischaemic region (3) lead to further release of cytokines, amplification of neuroinflammation and continued neuronal damage. In parallel, neurons release ‘help me’ signals (4) that activate astrocytes and microglia to release factors that protect neurons. MMPs, matrix metalloproteinases; TH1 cell, T helper 1 cell; TH17 cell, T helper 17 cell.
Over the past 40 years, trials of cytoprotective monotherapy have focused on pharmaceutical agents that target one component of the ischaemic cascade. However, such single-target therapeutics are likely to have a limited impact on the complex array of pathophysiological mechanisms unleashed by focal cerebral ischaemia. A more attractive prospect is the use of cytoprotective approaches that affect multiple aspects of the ischaemic cascade; for example, 3K3A-activated protein C acts on protease-activated receptor 1 to alter multiple downstream pathways involved in the ischaemic cascade, and nerinetide is an upstream inhibitor of excitotoxic glutamate signalling8,9. Hypothermia, although not a pharmacological therapy, is an example of an attractive multimodal approach that targets various components of the ischaemic cascade and that cytoprotective therapies could emulate. Hypothermia has not been successful in clinical trials owing to its adverse effects, such as pneumonia, and the need for anti-shivering medication10, but new approaches that achieve rapid reductions in temperature are focusing on focal brain cooling.
A second target for cytoprotective approaches is reperfusion injury, a mechanism that has long been of theoretical interest but is now clinically relevant in the era of reperfusion for AIS11. The concept of reperfusion injury is that the restoration of oxygenated blood flow into previously ischaemic brain tissue can have deleterious consequences in addition to the obvious benefits. For example, oxygenated blood can lead to the generation of reactive oxygen species, such as superoxide and peroxynitrite, that can overwhelm endogenous protective mechanisms, leading to tissue injury12. In addition, oxygenated blood can activate the immune system through the recruitment of leukocytes — activation of microglia can lead to tissue injury through the release of cytokines, complement and chemokines13, and the release of matrix metalloproteinases from neutrophils can lead to degradation of the extracellular matrix and tight junctions, contributing to BBB disruption, leakage and rupture14. The most severe consequence of this BBB disruption is haemorrhagic transformation in the ischaemic territory14,15. The later perfusion is restored in the ischaemic territory, the higher the risk of haemorrhagic transformation16. Finally, the reperfusion treatments can contribute to reperfusion injury themselves: tissue plasminogen activator (tPA) upregulates matrix metalloproteinases with the risk of BBB disruption, and thrombectomy can cause injury to the endothelial lining. These processes can initiate haemorrhagic transformation and lead to intracerebral haemorrhage.
A third cytoprotective strategy in AIS is the activation of endogenous collaterals in the brain to increase blood flow into the ischaemic region, thereby extending survival of the penumbra17. The extent and adequacy of collateral blood flow is one of the most important factors that determines the speed of progression towards irreversible injury and the extent of that injury in AIS. Therefore, enhancing collateral blood flow should be viewed as a valuable way to protect brain cells during ischaemia, especially soon after onset. Several approaches to increasing collateral blood flow are under development, including stimulation of the sphenopalatine ganglion that is close to gaining regulatory approval18.
A fourth potential cytoprotective strategy is the development of immunomodulators. Peripheral immune cells travel to the site of ischaemic injury and promote further damage through the release of cytokines and consequent amplification of pro-inflammatory microglia and astrocytes13. To date, no anti-inflammatory drugs have been proven to improve outcomes in stroke clinical trials. However, our understanding of how and when to modify the inflammatory response to produce beneficial effects needs further study. We have learned from clinical trials that broadly suppressing immune cell trafficking to the brain is not beneficial after stroke19 and that broadly suppressing the inflammatory response can even be harmful20. Approaches that target microglial activation are under development, as are applications of cell-based therapies that selectively downregulate pro-inflammatory signalling and upregulate anti-inflammatory signalling21.
No comments:
Post a Comment