Now we just need followup research in humans. WHOM IS GOING TO DO THAT? Specific names only.
Epicortical Brevetoxin Treatment Promotes Neural Repair and Functional Recovery after Ischemic Stroke
by
 Erica Sequeira 1,
Erica Sequeira 1,  Marsha L. Pierce 1, Dina Akasheh 1, Stacey Sellers 1, William H. Gerwick 2
Marsha L. Pierce 1, Dina Akasheh 1, Stacey Sellers 1, William H. Gerwick 2 , Daniel G. Baden 3 and Thomas F. Murray 1,*
, Daniel G. Baden 3 and Thomas F. Murray 1,*
Erica Sequeira 1, Marsha L. Pierce 1, Dina Akasheh 1, Stacey Sellers 1, William H. Gerwick 2, Daniel G. Baden 3 and Thomas F. Murray 1,*
1
Department of Pharmacology and Neuroscience, Creighton University, Omaha, NE 68123, USA
2
Center for Marine Biotechnology & Biomedicine, Scripps Institution of Oceanography, San Diego, La Jolla, CA 92093, USA
3
Center for Marine Science University of North Carolina Wilmington, Wilmington, NC 28409, USA
*
Author to whom correspondence should be addressed.
Mar. Drugs 2020, 18(7), 374; https://doi.org/10.3390/md18070374
Received: 25 June 2020 / Revised: 17 July 2020 / Accepted: 17 July 2020 / Published: 21 July 2020
(This article belongs to the Special Issue Marine Natural Products against Brain Diseases and Injuries)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Emerging literature suggests that after a stroke, the peri-infarct
region exhibits dynamic changes in excitability. In rodent stroke
models, treatments that enhance excitability in the peri-infarct
cerebral cortex promote motor recovery. This increase in cortical
excitability and plasticity is opposed by increases in tonic GABAergic
inhibition in the peri-infarct zone beginning three days after a stroke
in a mouse model. Maintenance of a favorable excitatory–inhibitory
balance promoting cerebrocortical excitability could potentially improve
recovery. Brevetoxin-2 (PbTx-2) is a voltage-gated sodium channel
(VGSC) gating modifier that increases intracellular sodium ([Na+]i), upregulates N-methyl-D-aspartate receptor (NMDAR) channel activity and engages downstream calcium (Ca2+)
signaling pathways. In immature cerebrocortical neurons, PbTx-2
promoted neuronal structural plasticity by increasing neurite outgrowth,
dendritogenesis and synaptogenesis. We hypothesized that PbTx-2 may
promote excitability and structural remodeling in the peri-infarct
region, leading to improved functional outcomes following a stroke. We
tested this hypothesis using epicortical application of PbTx-2 after a
photothrombotic stroke in mice. We show that PbTx-2 enhanced the
dendritic arborization and synapse density of cortical layer V pyramidal
neurons in the peri-infarct cortex. PbTx-2 also produced a robust
improvement of motor recovery. These results suggest a novel
pharmacologic approach to mimic activity-dependent recovery from stroke.
1. Introduction
Ischemic stroke is a common neurological disorder and major cause of long-term disability worldwide [1].
Currently, tissue plasminogen activator (tPA) is the only FDA-approved
pharmacologic treatment for ischemic or thrombotic stroke, which carries
the risk of producing an intracerebral hemorrhage [2,3,4].
Although this pharmacologic advancement of acute care has resulted in a
decline in mortality rate, it has produced a greater number of disabled
survivors. Soon after stroke onset, oxygen-deprived neurons in the
infarct core cease to function while tissue in the surrounding
peri-infarct region remain viable but compromised [5].
Previous work has suggested parallels between plasticity mechanisms in
the developing brain and those occurring in the adult brain after a
stroke event [6,7,8,9,10].
Neuronal circuits do undergo limited re-mapping and reorganization
after stroke, and these repair processes are associated with
neurogenesis, dentritogenesis, synaptogenesis, axonal sprouting and
rewiring of cortical networks in the peri-infarct tissue [11].
However, this spontaneous reorganization only partially restores motor
function. To more fully regain recovery of motor function, additional
pharmacologic manipulations in the peri-infarct are required.
Glutamate-mediated
excitotoxicity has been shown to contribute to ischemic cell death due
to failure of ionic homeostasis and a sustained elevation of
intracellular calcium concentration [12].
Following the excitotoxicity-induced acute insult, there is a period of
recovery with characteristic heightened neuroplasticity in the
peri-infarct tissue [13].
It is therefore critical that pharmacologic treatments to promote
recovery be administered subsequent to the acute phase of the stroke. In
rodent stoke models, pharmacologic and genetic strategies that enhance
neuronal excitability in the peri-infarct cortex adjacent to the stroke
promote motor recovery [14]. These mechanisms that enhance neuronal plasticity are similar to those involved in learning and memory [13].
In this regard, it is noteworthy that N-methyl-D-aspartate ionotropic
glutamate receptors (NMDARs) are crucial in activity-dependent synaptic
changes and in learning and memory.
Previous research has shown that changes in intracellular sodium concentration ([Na+]i)
produced in the soma and dendrites as a result of neuronal activity may
act as a signaling molecule and play a role in activity-dependent
synaptic plasticity. Synaptic stimulation elevates [Na+]i to 10 mm in dendrites and up to 35–40 mm in dendritic spines [15]. In hippocampal neurons, such intracellular [Na+]
increments have been demonstrated to increase NMDAR-mediated whole-cell
currents and NMDAR single-channel activity by increasing both channel
open probability and mean open time [16].
Brevetoxins (PbTx-1 to PbTx-10) are potent lipid soluble polyether neurotoxins produced by the marine dinoflagellate Karenia brevis [17].
PbTx-2 interacts with neurotoxin site 5 on the α subunit of
voltage-gated sodium channels (VGSCs), and augments sodium influx by
inhibiting channel inactivation and shifting the activation potential to
more negative values [18].
Src kinases are widely expressed in the brain and regulate activities
of ion channels such as NMDARs. Phosphorylation of NMDAR tyrosine
residues by Src facilitates the binding of Na+ to NMDAR and exerts a regulatory effect on NMDAR signaling [19].
Single-channel currents recorded from cell-attached patches on
cerebrocortical neurons indicate that PbTx-2 upregulates NMDAR
whole-cell currents by increasing mean open time and probability without
affecting the resting membrane potential [20]. This upregulation is attributed to the coincident elevation of intracellular [Na+] and Src kinase activation [21]. PbTx-2 treatment of cerebrocortical neuron cultures robustly potentiated NMDAR-mediated calcium influx (Ca2+) [20].
In immature cerebrocortical neurons, PbTx-2 treatment enhanced neurite
outgrowth, dendritic arborization, synaptogenesis and filopodia
formation and maturation [22].
In addition, PbTx-2 exposure engaged downstream activity-dependent
mechanisms involved in neuronal growth and survival such as Ca2+-calmodulin
kinases (CaMKs), extracellular signal-regulated kinase (ERK), cAMP
response element binding protein (CREB) and brain-derived neurotrophic
factor (BDNF) signaling pathways [22].
PbTx-2 exhibited a characteristic bidirectional concentration–response
profile similar to that of NMDA since an optimal window for [Ca2+]i is required for neurite extension and branching [23].
Inasmuch as the mechanisms involved in repair processes after stroke
are similar to those regulating neuronal development, we hypothesized
that PbTx-2 may augment recovery following ischemic stroke.
We
therefore explored neurohistochemical and functional outcomes of
administration of PbTx-2 during the recovery phase after stroke. To
assess neurohistochemical changes, we imaged neurons in the peri-infarct
cortex to assess dendritic arborization and synaptic density. In
humans, long-term disabilities related to stroke often include
impairments in feeding, coordination and gait. To examine impairment and
recovery, we utilized a catwalk test to examine gross motor gait, a
pasta matrix reach task to assess fine-motor skills (feeding and
coordination) and a foot fault task to examine coordination and gait. An
emerging strategy in stroke therapy is the direct application of
treatments to the stroke lesion [24,25,26].
Accordingly, we mixed PbTx-2 in a hydrogel composed of thiol-modified
hyaluronan and polyethylene glycol diacrylate and this composite was
deposited epicortically directly above the stroke cavity. We demonstrate
that epicortical application of PbTx-2 at five-days post-infarct
enhances neuronal repair and improves functional outcomes.
2. Results
2.1. PbTx-2 Enhances Neuronal Structural Plasticity in the Peri-Infarct Region as Revealed by Increased Dendritic Arbor Complexity and Synapse Formation
Using 2- to 4-month-old male yellow
fluorescent protein (YFP) line-H transgenic mice, we produced unilateral
photothrombotic strokes by providing an intraperitoneal injection of a
light sensitive dye followed by exposure of the motor cortex region to a
cold source of light. Mice were then allowed to recover in home cages
for five days (Figure 1).
On day 5, animals were treated with the vehicle or PbTx-2 and
subsequently sacrificed on day 6 to examine neuronal structural changes
in the peri-infarct region.
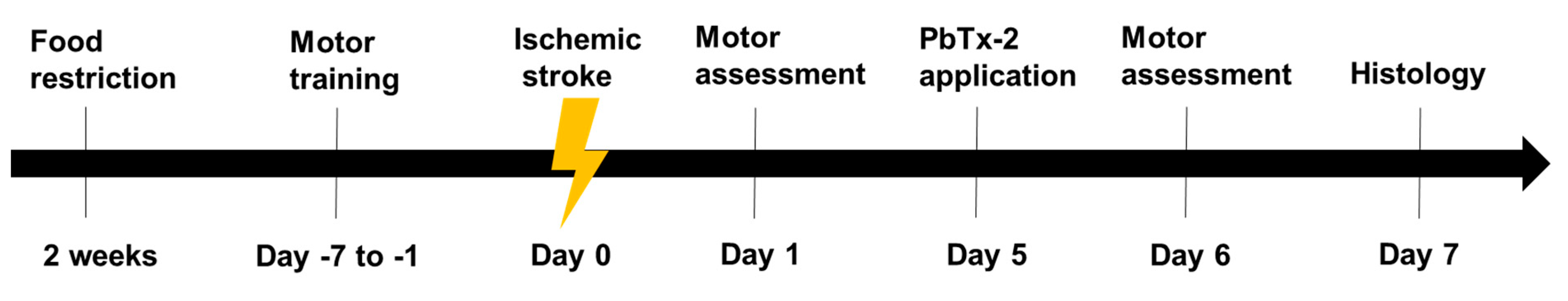
Figure 1.
Experimental timeline: Animals were food-restricted to 85% of their body
weight for two weeks prior to training for pasta matrix reach task.
Animals were trained to perform pasta matrix reach task and foot fault
task prior to inducing stroke to obtain baseline values. A focal lesion
was induced in the motor cortex region by photothrombosis on day 0.
Animals were randomly divided into PbTx-2 or vehicle treatment groups.
On day 5, PbTx-2 mice were treated with 3, 10, 100, 1000 or 3000 pmol
PbTx-2, whereas vehicle-treated animals were given hydrogel alone.
Further, on day 1 (post-stroke) and day 6 (post-treatment), animals were
assessed for the number of pasta pieces retrieved and percentage of
foot faults. On day 6, animals were sacrificed, and brains were isolated
for histological analysis of surviving neurons in the peri-infarct
region.
We performed cresyl violet staining to
evaluate the extent of the photothrombotic stroke volume and the effect
of epicortical application of PbTx-2 on the same (Figure 2A).
Coronal sections of 100 μm were collected using a vibratome (Leica VT
1200S). For the infarct volume measurement, brain sections were stained
with 0.5% cresyl violet and images were then acquired using a
bright-field microscope. The areas of infarct were delineated and
quantified using the Image J software (NIH) and infarct volume was
calculated by summation of the lesion areas of all sections and
integrated by the thickness of the section (Figure 2B). Infarct volumes did not vary significantly between vehicle- and PbTx-2-treated mice (Figure 2C).
These results indicate that the timing of PbTx-2 treatment utilized was
appropriate for the assessment of promoting functional recovery
inasmuch as these treatments did not affect stroke volume.
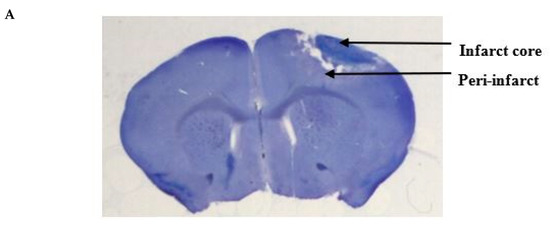
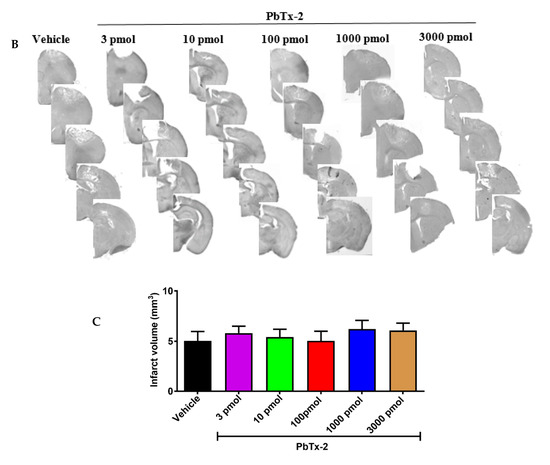
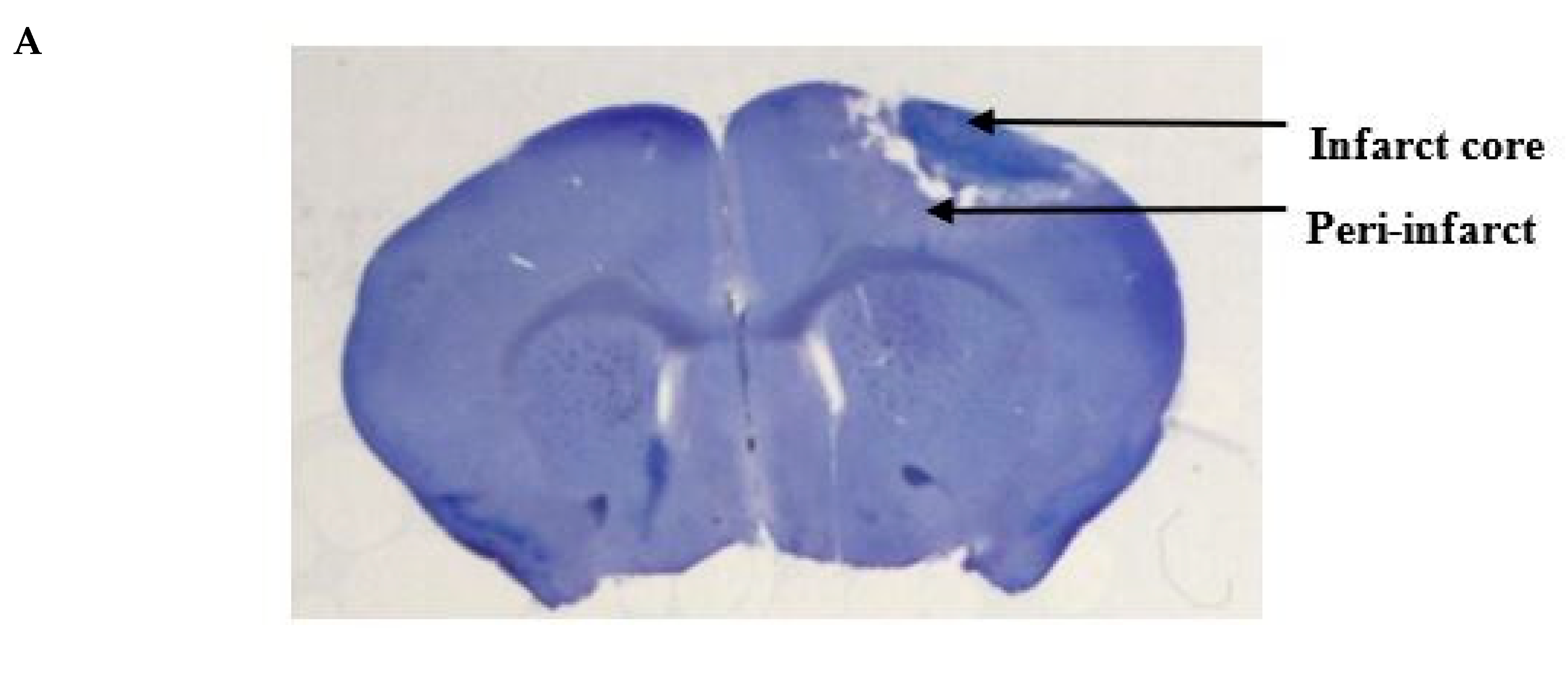
Figure 2.
Histologic assessments after stroke. (A) Representative cresyl violet image showing the infarct and the peri-infarct region. (B) Representative cresyl violet stained-sections of vehicle, 3, 10, 100, 1000 and 3000 pmol PbTx-2, respectively. (C)
Quantification of infarct volume. No significant differences in infarct
size were detected (one-way ANOVA followed by Dunnett’s post hoc test, 3
pmol PbTx-2 effect, p = 0.993; 10 pmol PbTx-2 effect, p = 0.907; 100 pmol PbTx-2 effect, p > 0.999; 1000 pmol PbTx-2 effect, p = 0.873; 3000 pmol PbTx-2 effect, p = 0.975). Data shown are mean ± SEM of 2 to 7 brains.
Dendritic injury is a pathologic hallmark of excitotoxicity inasmuch as NMDARs are predominantly localized on dendrites [27,28].
Stroke caused the deterioration of neurons in the infarct core that
could be readily distinguished from the adjacent surviving peri-infarct
region. Confocal images of layer V YFP-labelled pyramidal neurons in the
peri-infarct region were obtained and 3D morphological analysis of the
arbor complexity with a defined algorithm was performed using the Imaris
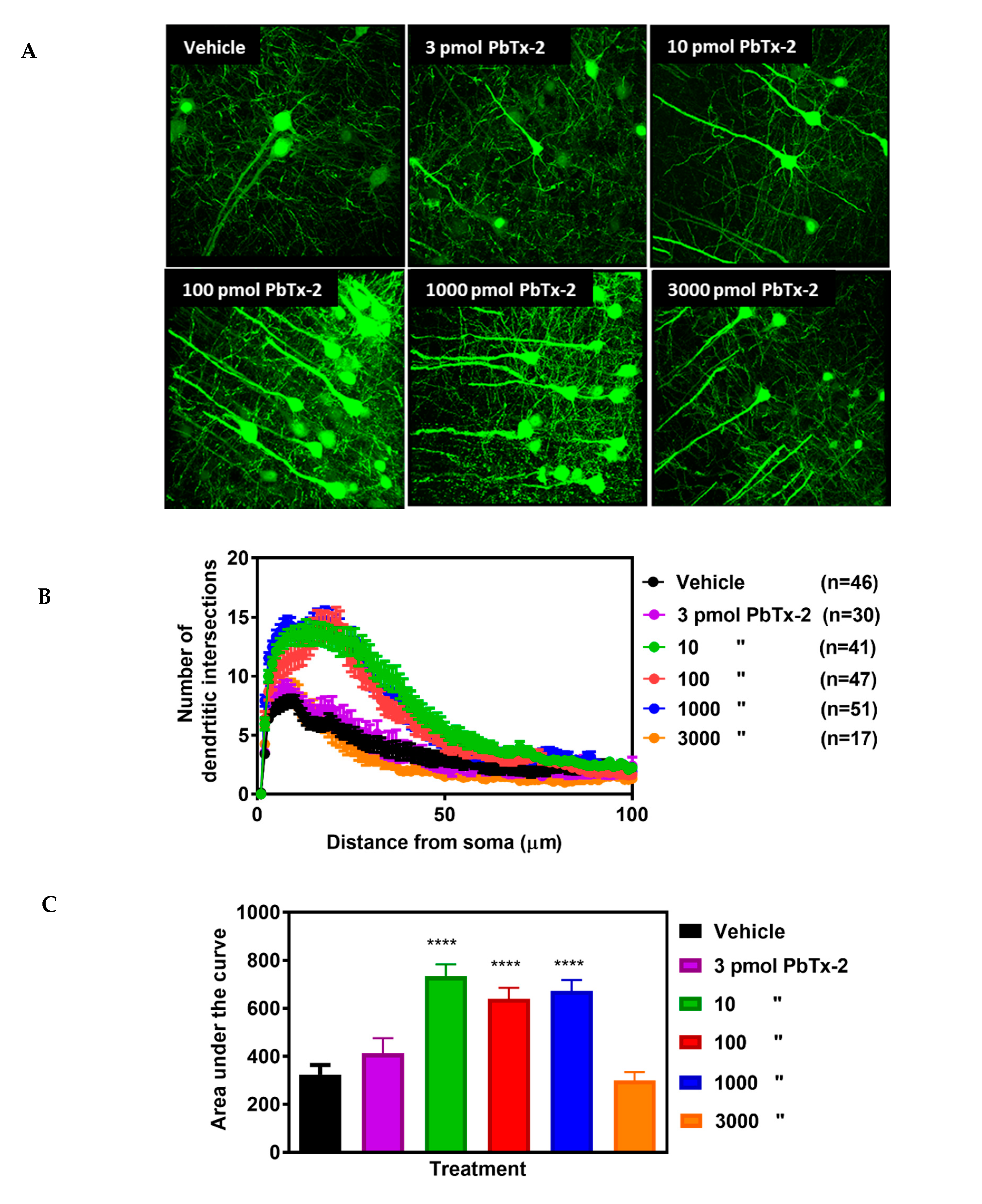
image analysis software (Figure 3A).
In vehicle-treated animals, there was a gradual increase in branching
complexity moving away from the soma, reaching a maximum of 8 ± 0.5
intersections per neuron, and then progressively declining beginning at
approximately 10 µm from the soma (Figure 3B).
Doses of 10, 100 and 1000 pmols of PbTx-2 produced a 2-fold increase in
the expansion of dendritic arbors of neurons in the peri-infarct
cortex, and a rightward shift in the Sholl plot as compared with the
vehicle-treated control mice (Figure 3B).
An AUC analysis of Sholl data showed a significant increase in
dendritic complexity following the 10, 100 and 1000 pmol doses of PbTx-2
compared with vehicle controls. The PbTx-2 effect on dendritic
complexity displayed a bidirectional profile as shown by a lack of
effect on the expansion of dendritic arbors at the 3 and 3000 pmol doses
(one-way ANOVA followed by Dunnett’s post hoc test, **** p < 0.0001; 3 pmol PbTx-2 effect, p = 0.579; 3000 pmol PbTx-2 effect, p = 0.998; n = 17 to 51 neurons; Figure 3C).
Given our previous demonstration of bidirectional PBTx-2
concentration–response profiles in cerebrocortical neurons similar to
that of NMDA, a possible explanation for PbTx-2’s bidirectional profile
herein is the underlying NMDAR-dependent mechanism of action [23].
Previous studies have established an inverted-U concentration–response
profile for the relationship between NMDAR and neuronal survival, where
too little or too large activation of NMDARs can respectively diminish
neuronal growth or cause cell death [29].
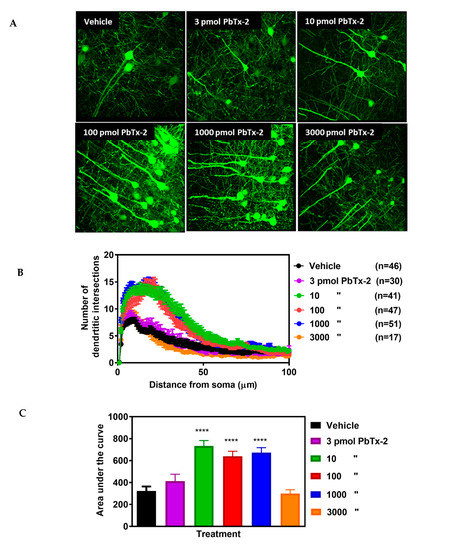
Figure 3.
Effect of PbTx-2 on dendritic arborization in the peri-infarct region. (A) Representative images of PbTx-2-induced dendritic arborization in the peri-infarct region (Scale bar: 30 μm). (B) Sholl analysis to quantify dendritic arbor complexity. (C)
Area under the curve (AUC) analysis of Sholl data, PbTx-2 at 10, 100
and 1000 pmol doses enhanced dendritic arbor complexity in the
peri-infarct site as compared with the vehicle-treated animals (one-way
ANOVA followed by Dunnett’s post hoc test, **** p
< 0.0001). However, the 3 and 3000 pmol doses of PbTx-2-treated mice
were without effect on the expansion of dendritic arbors (3 pmol PbTx-2
effect, p = 0.579; 3000 pmol PbTx-2 effect, p = 0.998). Each bar represents the mean ± SEM of 17–51 neurons.
Next, we examined the influence of PbTx-2 on
synapse formation in the peri-infarct cortex region. Confocal images of
YFP-labelled neurites in the peri-infarct region were obtained and
analyzed using a spot detection algorithm again using the Imaris image
analysis software (Figure 4A).
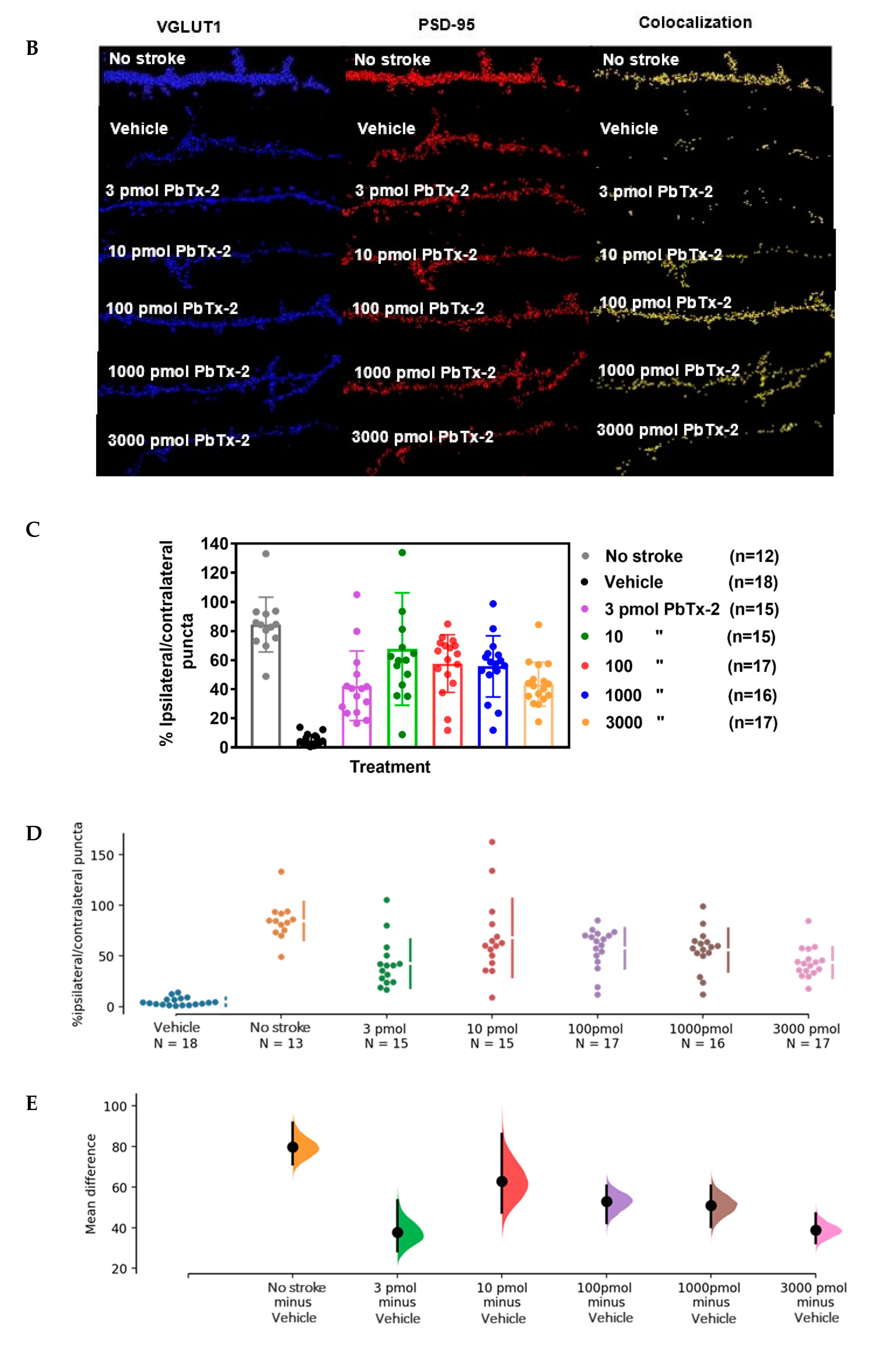
Antibodies against VGLUT1 (presynaptic marker) and PSD-95 (postsynaptic
marker) were used to quantify synapse density, as revealed by
colocalized fluorescent puncta (Figure 4B).
We obtained the ratio of synaptic puncta on the ipsilesional side to
that of the contralesional side to correct for between animal variation.
Dose–response analysis of the effect of PbTx-2 on synapse density
indicated that 3, 10, 100 and 1000 and 3000 pmol PbTx-2 produced a
significant increase in synapse formation (number of puncta per length
of neurite) compared with the vehicle-treated animals (one-way ANOVA
followed by Dunnett’s post hoc test, **** p < 0.0001; n= 12 to18 neurons; Figure 4C).
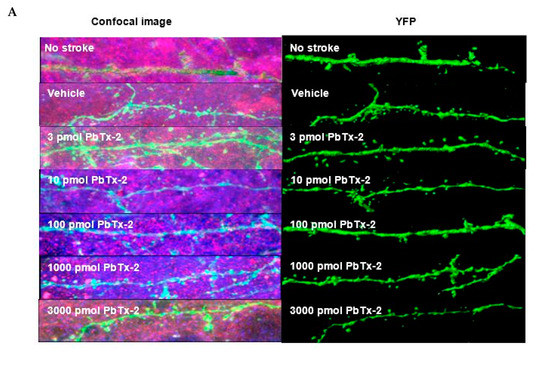
Figure 4.
Effect of PbTx-2 on excitatory synapse density in peri-infarct region. (A)
Representative images of double-immunostained YFP expressing neurites
in the peri-infarct region at day 6 obtained from a confocal microscope.
(B) Antibodies against VGLUT1 (presynaptic marker/blue) and
PSD-95/red (postsynaptic marker) were used to quantify synapse density,
as indicated by colocalized fluorescent puncta (yellow), scale bar: 5
µm. (C) Quantification of colocalized fluorescent puncta using
Imaris image analysis; 3, 10, 100, 1000 and 3000 pmol PbTx-2 doses
enhanced synapse density in the peri-infarct region (one-way ANOVA
followed by Dunnett’s post hoc test, **** p < 0.0001). (D)
Gardner–Altman mean comparison plots showing PbTx-2-induced increments
in synapse density. Each plot contains comparisons for all
PbTx-2-treated groups, and each dot represents %
ipsilateral/contralateral puncta from an individual brain section. (E)
Gardner–Altman plot demonstrating effect size. The left ordinate axis
of the plot shows mean difference (MD) distribution between the
PbTx-2-treated and vehicle-treated groups; 3 pmol PbTx-2 MD = 37.6%, 95%
CI [28.3, 53.4]; 10 pmol PbTx-2: MD = 63%, 95% CI [47.4, 86.2]; 100
pmol PbTx-2: MD = 52.9%, 95% CI [42.3, 60.7]; 1000pmol PbTx-2: MD = 51%,
95% CI [40.3, 60.6]; 3000 pmol PbTx-2: MD = 38.7%, 95% CI [32.4, 47.0
All data are represented as mean ± SEM of 12–18 brain sections.
Further, to better visualize the PbTx-2 effect
size, we compared mean difference (MD) distribution between the
vehicle- and PbTx-2-treated groups using the Gardner–Altman mean
comparison plot that affords transparency of the effect size of
treatments [30]
(3 pmol PbTx-2 MD = 37.6%, 95% CI [28.3, 53.4]; 10 pmol PbTx-2: MD =
63%, 95% CI [47.4, 86.2]; 100 pmol PbTx-2: MD = 52.9%, 95% CI [42.3,
60.7]; 1000pmol PbTx-2: MD = 51%, 95% CI [40.3, 60.6]; 3000 pmol PbTx-2:
MD = 38.7%, 95% CI [32.4, 47.0]; Figure 4D,E).
The effect sizes and CIs are reported as: effect size [CI width—lower
bound, upper bound] and the narrowness of the confidence interval
represents the effect size precision.
2.2. PbTx-2 Promotes Recovery of Fine Motor Skills in Stroke Affected Mice
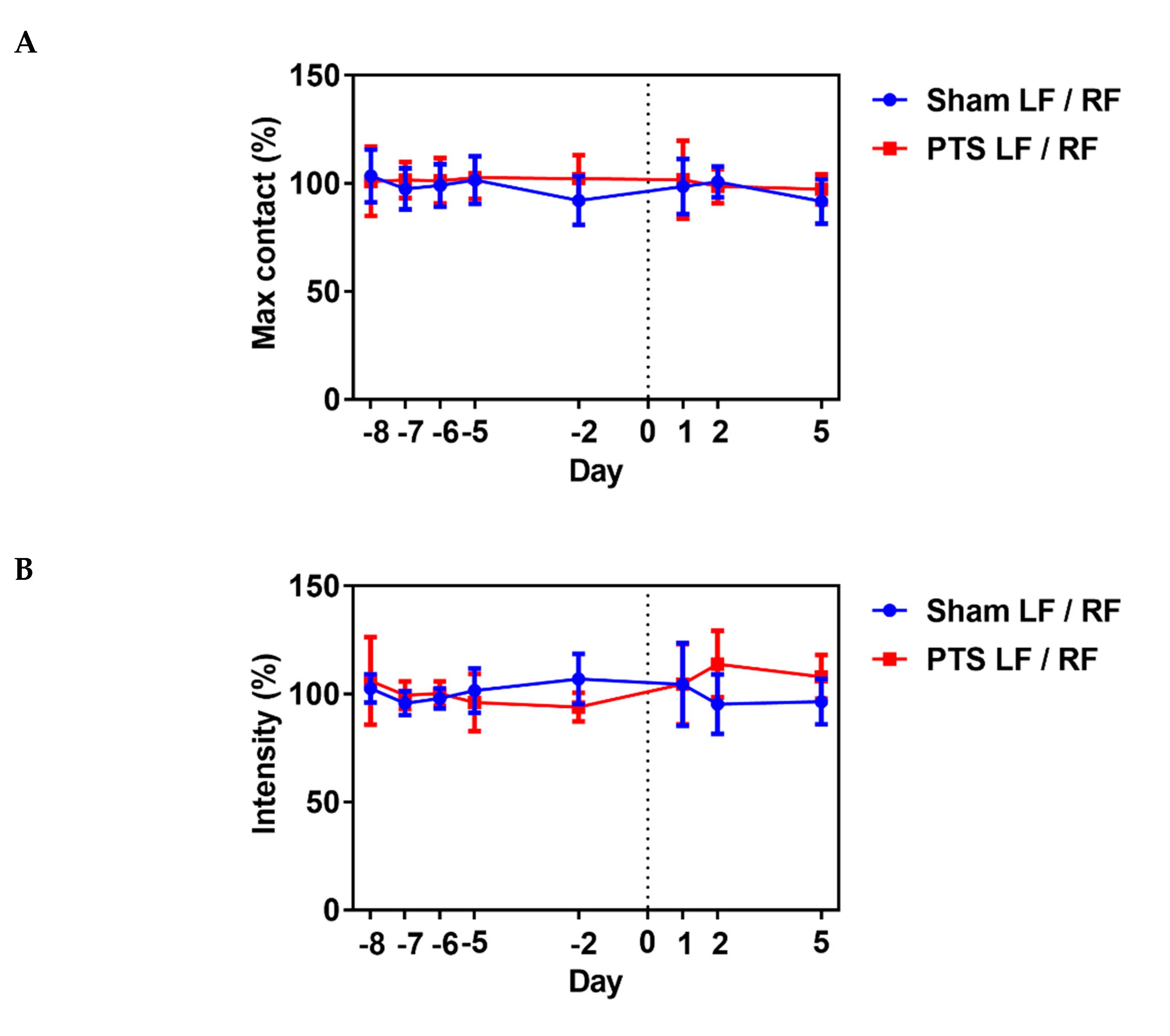
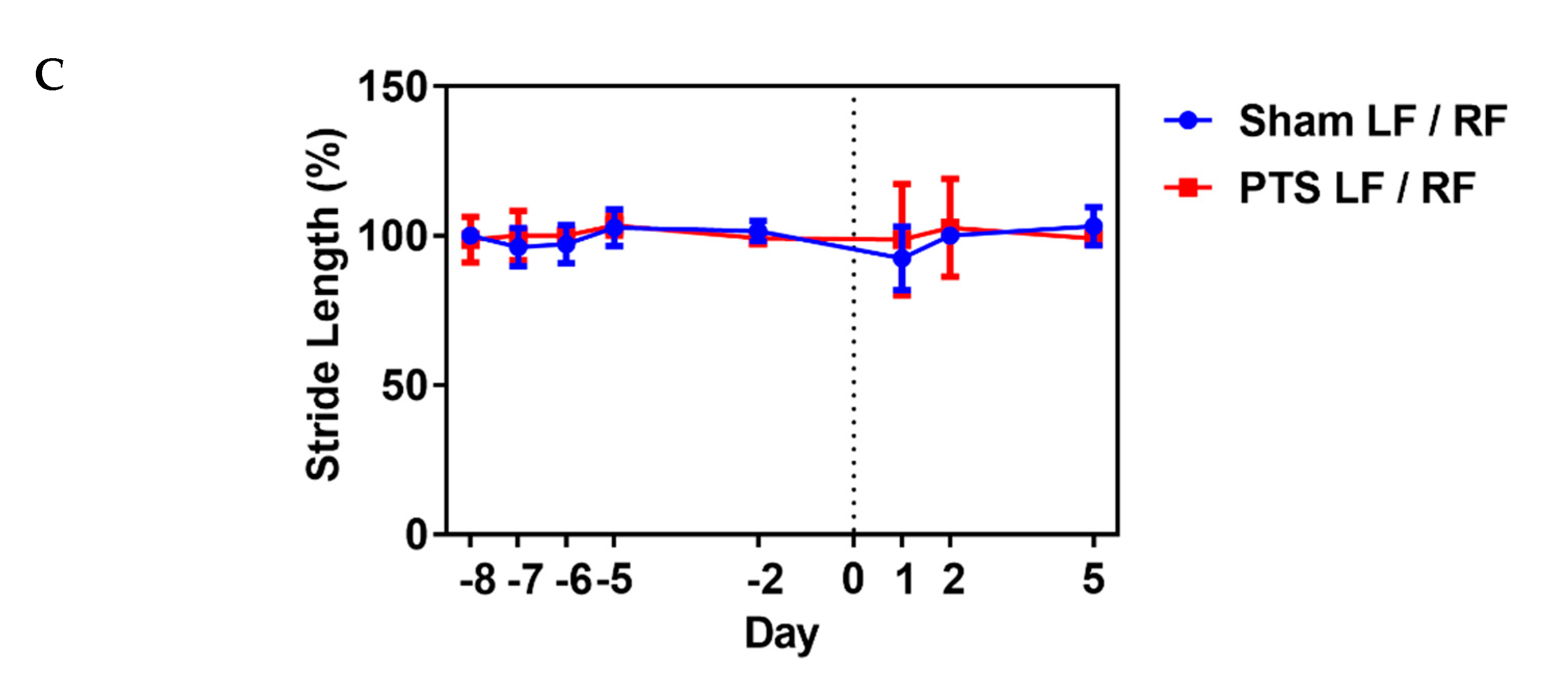
We
next assessed the influence of the photothrombotic focal stroke on
motor function and coordination by first performing a CatWalk gait
analysis. We examined contralateral forelimb function post-stroke as
motor disability in response to photothrombotic stroke [31].
We analyzed several gait parameters on days 1, 2 and 5 post-stroke and
compared the stroke ipsilateral left front (LF) paw to the contralateral
right front (RF) paw in each animal. No differences between the
sham-operated group and photothrombotic stroke (PTS) group were
detected. Shown are the results for parameters “max contact”,
“intensity” and “stride length” (unpaired two-tailed test: max contact
(%), p = 0.721; intensity (%), p = 0.987; stride length (%), p = 0.471; n = 9 mice; Figure 5).
These results demonstrated a lack of effect of the stroke on gait
parameters and that motor deficits might be confined to the digits of
the paw. We therefore next used a pasta matrix reach task and a foot
fault test to assess fine motor skills.
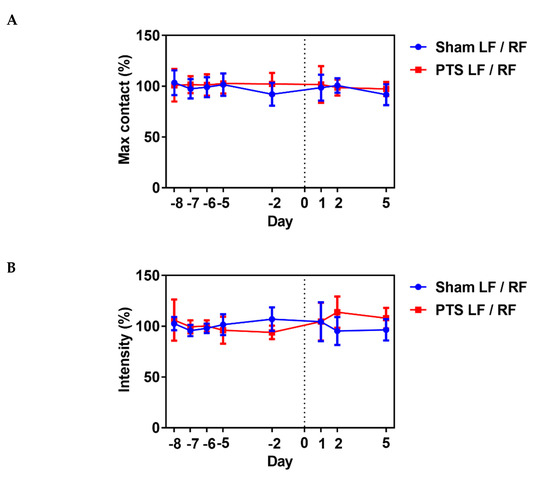
Figure 5.
Impact of photothrombotic stroke on gross motor function. Representative CatWalk parameters (A) max contact (%), (B) intensity (%) and (C)
stride length (%) analyzed before inducing photothrombotic stroke (PTS)
to obtain baseline and on days 1, 2 and 5 after stroke. We compared the
ipsilateral left front (LF) paw to the contralateral right front (RF)
paw. Gait parameters were not altered by photothrombotic stroke. Shown
are the results of unpaired two-tailed test (max contact (%), p = 0.721; intensity (%), p = 0.987; stride length (%), p = 0.471). All values are given as mean ± SEM (n = 9 mice).
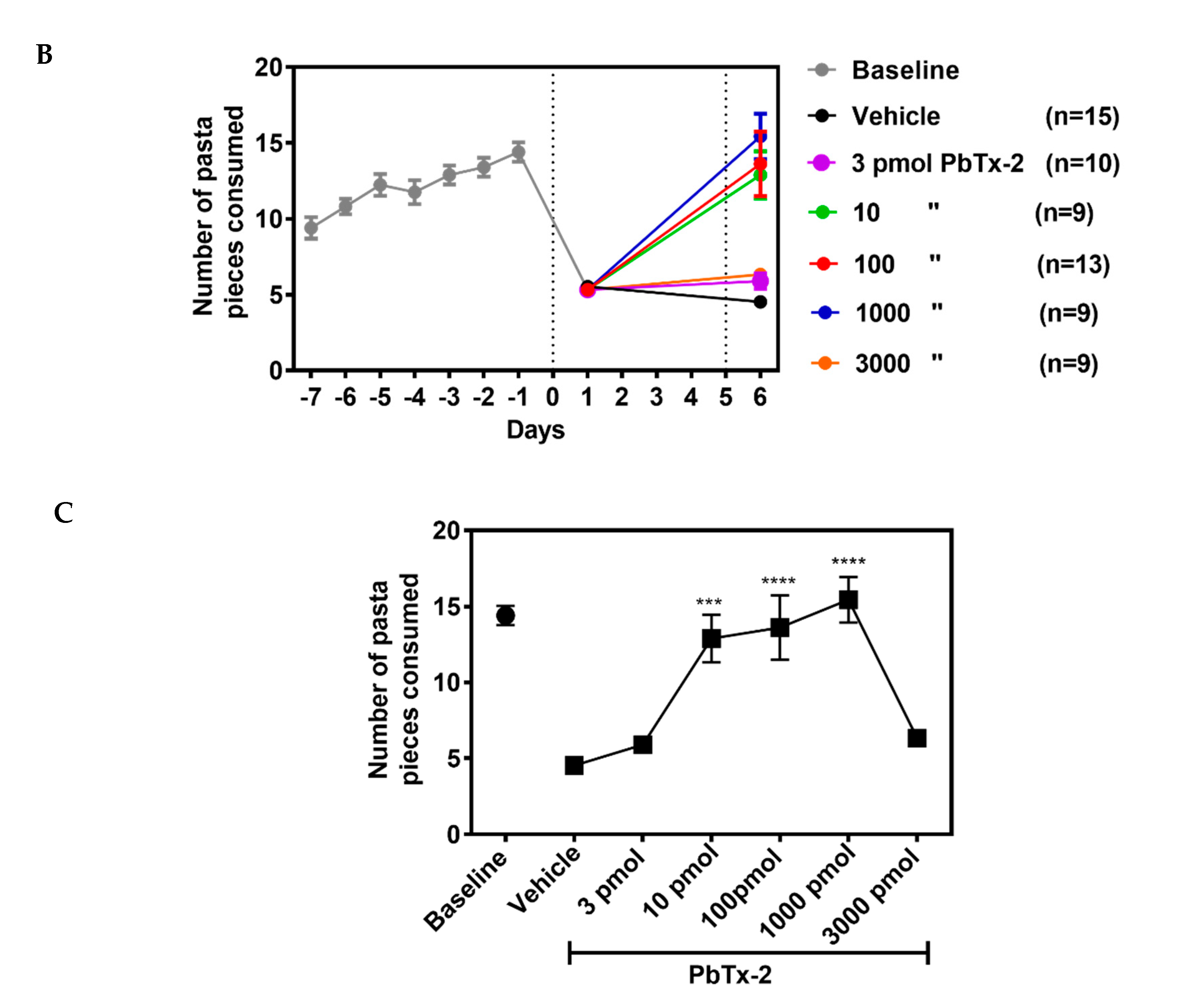
Rodents live in an environment that requires the use of a complex range of motor skills to gain access to food [32]. The pasta matrix reach task is one of the few motor tests that can measure skilled forepaw use [33]. Mice were trained for three days to grab a single piece of pasta to determine paw preference (Figure 6A), followed by seven days of pasta matrix training sessions to obtain baseline values before inducing stroke (Figure 6B).
On day 1, following the stroke, there was a significant decline in the
number of pasta pieces retrieved, indicating that the photothrombotic
stroke affected fine motor skills. On day 6, following the stroke,
animals treated with the 10, 100 and 1000 pmol doses of PbTx-2 exhibited
a significant increase in the number of pasta pieces retrieved as
compared with the vehicle-treated mice. The lowest and highest doses of
PbTx-2, 3 and 3000 pmol, were however without a significant effect
(one-way ANOVA followed by Dunnett’s post hoc test, **** p < 0.0001, *** p = 0.0002; 3 pmol PbTx-2 effect, p = 0.955; 3000 pmol PbTx-2 effect, p = 0.873; n = 9 to 15 mice; Figure 6).
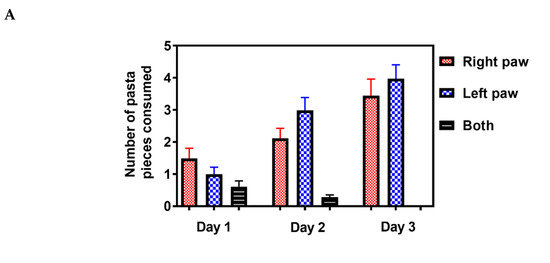
Figure 6.
Effect of PbTx-2 treatment on pasta matrix reach task. (A) Paw preference was determined using a single piece of pasta for 3 days. (B)
Animals were trained for 7 days to retrieve 25 pieces placed in a 5X5
matrix from their preferred paw. Day 0 represents the photothrombotic
procedure and at 1 day after the stroke, there was a significant
reduction in the number of pasta pieces retrieved, indicating motor
deficits induced by the insult. On day 6, after the stroke, the 10, 100
and 1000pmol PbTx-2-treated animals exhibited significant increases in
the number of pasta pieces retrieved as compared with the
vehicle-treated animals. The 3 and 3000 pmol PbTx-2 doses did not
facilitate functional recovery. (C) Quantification of PbTx-2
dose–response effects on motor recovery 6 days post-stroke (one-way
ANOVA followed by Dunnett’s post hoc test, **** p < 0.0001, ***p = 0.0002; 3 pmol PbTx-2 effect, p = 0.955; 3000 pmol PbTx-2 effect, p
= 0.873). All data are represented as mean ± SEM (n = 9 to 15 mice).
Data points without error bars are due to the error bar being smaller
than the symbol.
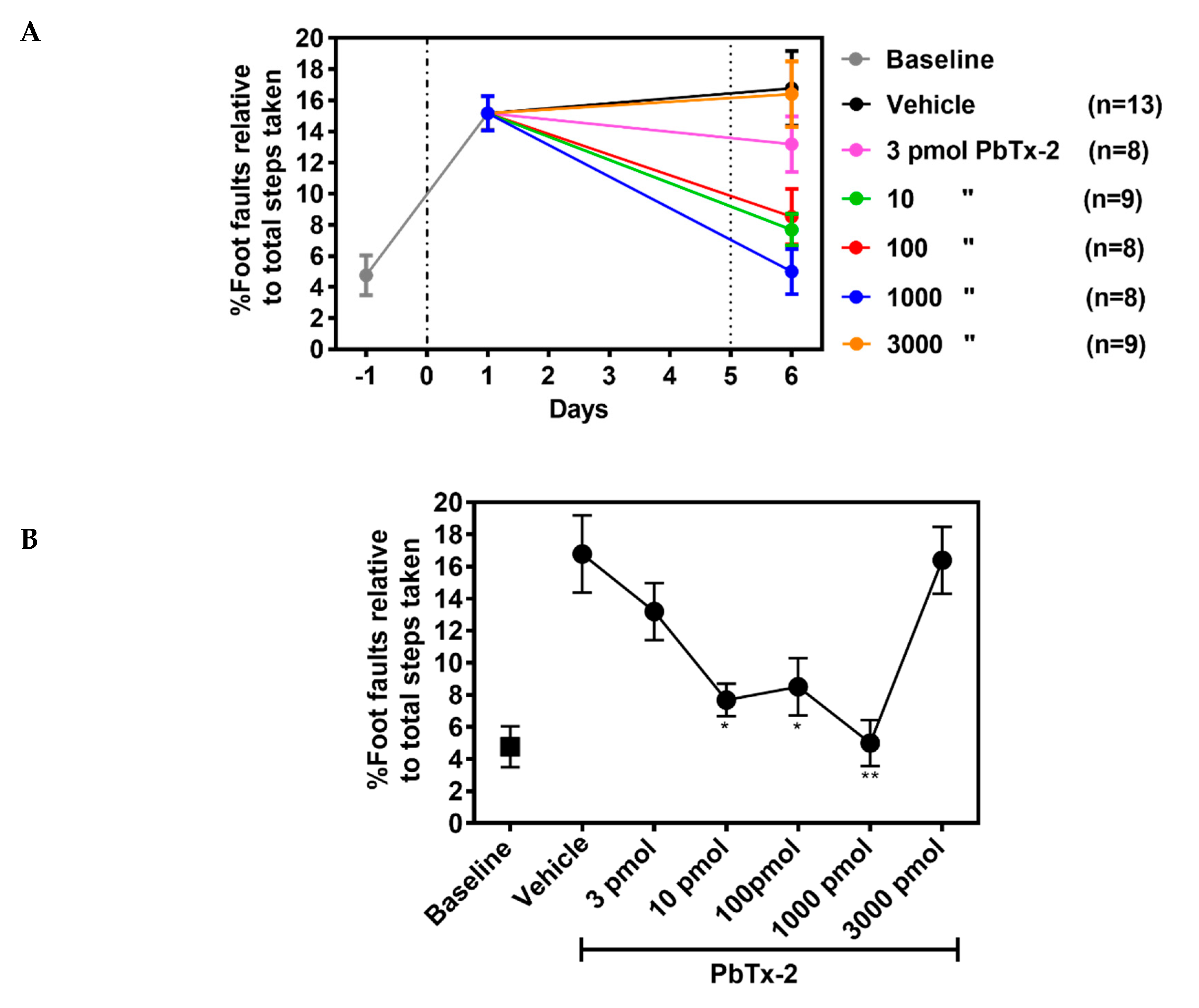
To further confirm the influence of PbTx-2 on
functional recovery, we used a foot fault task that represents a
sensitive method for detecting motor deficits of limb functioning and
placement during locomotion. Animals without a stroke should place their
paws precisely on the wire frame and demonstrate few to zero foot
faults [33].
The photothrombotic stroke in the forelimb motor cortex produced a
significant increase in the percentage foot faults on day 1 after the
insult, indicating a disruption of limb function and placement (Figure 7A).
At PbTx-2 doses of 10, 100 and 1000 pmols, treated animals displayed
significant improvements in percentage of foot faults as compared with
the vehicle-treated mice. Again, the 3 and 3000 pmol doses of PbTx-2 did
not promote functional recovery (one-way ANOVA followed by Dunnett’s
post hoc test, 10 pmol PbTx-2 effect; * p = 0.023, 100 pmol PbTx-2 effect; * p = 0.027, 1000 pmol PbTx-2 effect; ** p = 0.001; 3 pmol PbTx-2 effect, p = 0.703; 3000 pmol PbTx-2 effect, p > 0.999; n = 9 to 13 mice; Figure 7B).
These data establish that treatment with the 10, 100 and 1000 pmol
doses of PbTx-2 on day 5 post-stroke resulted in functional recovery of
fine motor skills in both the pasta matrix handling and foot fault tasks
as compared with the vehicle control treatments.
Figure 7.
Effect of PbTx-2 treatment on foot fault task. (A) Animals were
trained to walk on an elevated grid prior to photothrombotic stroke to
obtain baseline values. On day 1 after inducing stroke, a significant
increase in percentage of foot faults was observed. The 10, 100 and 1000
pmol doses of PbTx-2 produced significant improvement in percentage of
foot faults as compared with the vehicle-treated animals. Alternatively,
the 3 and 3000 pmol doses of PbTx-2 did not aid recovery. (B)
Quantification of PbTx-2 dose–response effects on motor recovery 6 days
post-stroke (one-way ANOVA followed by Dunnett’s post hoc test, 10 pmol
PbTx-2 effect, * p = 0.023; 100 pmol PbTx-2 effect, * p = 0.027; 1000 pmol PbTx-2 effect, ** p = 0.001; 3 pmol PbTx-2 effect, p = 0.703; 3000 pmol PbTx-2 effect, p > 0.999). All data points are represented as mean ± SEM (n = 9 to 13 mice).
3. Discussion
Here,
we investigated the effect of PbTx-2 on neuroplasticity in the
peri-infarct cortex and associated motor functions in a murine model of
stroke. The main findings of this study are: 1. epicortical application
of PbTx-2 at the stroke site produced a 2-fold increase in dendritic
arborization and increased synaptogenesis in the peri-infarct cortex, 2.
photothrombotic stroke in the forelimb motor cortex produced functional
deficits that were confined to the digits of the paw, 3. PbTx-2 doses
of 10, 100 and 1000 pmols produced dramatic improvements in functional
recovery toward pre-stroke controls as measured by an increase in the
number of pasta pieces retrieved or decreased percentage of foot faults
and 4. PbTx-2 displayed bidirectional dose–response profiles where the 3
and 3000 pmol doses did not affect neurite outgrowth or motor
functional recovery, consistent with these effects being mediated
through NMDARs.
VGSCs play a fundamental role in electrical signaling of the nervous system and action potential generation [34]. Two-photon imaging studies show that synaptic stimulation leads to transient increases in [Na+]i in postsynaptic spines and dendrites [15]. This suggests that [Na+]i
may function as a signaling molecule and play a role in
activity-dependent synaptic plasticity. PbTx-2, a VGSC gating modifier,
augments NMDA receptor signaling through coincidence of an elevation of
[Na+]i and Src kinase activity [21].
A previous report suggested that PbTx-2-mediated activation of sodium
channels was associated with enhancement of NMDA-induced Ca2+
influx, accelerated spine formation and maturation, increased dendritic
arbor elaboration and increased synaptogenesis in developing
cerebrocortical neurons [22]. The cell signaling mechanisms underlying these responses involved PbTx-2-induced increase in intracellular Ca2+ with attendant phosphorylation of Ca2+-dependent
molecules including CaMKI, CaMKII and CREB that play essential roles in
neuronal growth and survival. BDNF is an activity-dependent
neurotrophic factor that mediates neuroplasticity and is regulated by
CREB-dependent mechanisms [35],
and PbTx-2 exposure also increased the surface expression of
BDNF-tropomyosin-related kinase B receptors in cerebrocortical neurons [22].
Glutamate
plays an essential role in mediating excitatory neurotransmission in
the central nervous system and is vital for synaptic plasticity. After
an ischemic stroke, however, glutamate accumulation leads to
excitotoxicity due to over-activation of NMDARs and neuronal death [36,37].
Interestingly, NMDAR antagonists failed clinically to show
neuroprotective effects and, in some cases, worsened stroke outcomes in
patients [38,39,40,41].
Hence, blocking NMDARs subsequent to a stroke is detrimental inasmuch
as glutamate signaling through NMDARs contributes to neuronal survival.
This influence of glutamate on NMDARs to promote neuronal survival
displays an inverted U-shaped concentration–response curve, where too
little or excessive activation of NMDARs are detrimental [42].
Neuronal excitability after a stroke exhibits distinct phases during stroke progression and recovery [43].
In the acute phase, excessive glutamatergic activity produces
excitotoxicity and is deleterious. During the subsequent chronic phase
however, glutamatergic excitability in the peri-infarct cortex is
correlated with neuronal repair and recovery [43].
Therefore, enhancing cortical excitability too early after stroke may
further increase neuronal death. This inflection point from the acute
excitotoxic to chronic recovery phase occurs three days post-stroke in
mice [14].
In the present study, we therefore selected the time point of five days
after stroke for the epicortical treatments. Stroke recovery has been
associated with dramatic spine plasticity in the peri-infarct cortex and
with an increase in dendritic spine density over baseline values in
some regions [11].
This influence of glutamatergic signaling is opposed by a marked
increase in extracellular γ-aminobutyric acid (GABA) levels due to the
loss of GABA transporter GAT-3 [14]. Notably, administration of L655,708, a benzodiazepine inverse agonist specific for extrasynaptic GABAA receptors, produced a rapid and sustained improvement in functional recovery in mice following a photothrombotic stroke [14].
Hence, counteracting the hypo-excitability caused by heightened
GABAergic inhibition could potentially promote recovery when initiated
during the chronic phase. The present results using the sodium channel
gating modifier brevetoxin may provide an additional approach for
enhancing brain excitability during the period of recovery and
reorganization to promote neural repair.
We
selected the photothrombotic stroke model because it produces a
localized infarct that permits a detailed analysis of neuronal
structural plasticity and functional recovery [44].
The effect of photothrombotic stroke on forelimb fine motor deficits,
as assessed by the pasta matrix reach and foot fault tasks, appeared to
be both pervasive and persistent since at six days post-stroke, the
vehicle-treated animals displayed profound deficits in task performance.
We found that a single epicortical PbTx-2 treatment applied five days
post-stroke was sufficient to promote functional recovery and that these
beneficial effects were paralleled by PbTx-2-induced increases in
dendritic arbor complexity and synaptic density in the peri-infarct
cortex. These actions of PbTx-2 on neuronal plasticity and functional
recovery both showed inverted-U dose–response curves. We have shown
previously that the in vitro effect of PbTx-2 on neurite outgrowth in
cerebrocortical neurons exhibited a bidirectional concentration–response
(inverted-U) profile and that this effect was primarily dependent on
NMDARs [20].
Similarly, the effects of PbTx-2 on dendritic arborization and
synaptogenesis in cerebrocortical neurons displayed bidirectional
concentration–response profiles [22]. The inverted-U model for the relationship between NMDAR activity and neuronal survival and growth is well established [29].
The inverted-U dose–response effects of PbTx-2 on neuronal plasticity
in the peri-infarct cortex observed in the present study are consonant
with those of a previous report that neuronal activity affects
structural plasticity in vivo through NMDAR-triggered intracellular
signaling events [45].
It is therefore reasonable to posit that the effects of epicortical
PbTx-2 on structural plasticity and post-stroke functional recovery are
the result of elevated [Na+]i and enhanced NMDAR function.
Our
results demonstrate impairment in forelimb fine motor control in mice
after a photothrombotic stroke and reversal of these deficits by PbTx-2
treatment. These data suggest that stroke-induced motor deficits might
be particularly responsive to augmented cortical excitability during the
recovery phase of stroke. Currently, the only clinical treatment
following a stroke is tissue plasminogen activator (tPA) which must be
administered within the first few hours post-stroke. Considering that
occupational and physical therapy are the standard of care for stroke
recovery, our results suggest that sodium channel gating modifiers may
represent a novel pharmacotherapy to accelerate recovery. This new
strategy to enhance cortical excitability during the delayed time frame
important for neural repair and recovery may hold promise for reducing
the severity of stroke disability.
No comments:
Post a Comment