No clue.
Research Progress on the Mechanism of Mitochondrial Autophagy in Cerebral Stroke
 Li Lei1†,
Li Lei1†,  Tao Li
Tao Li- 1Department of Neurosurgery, The First People’s Hospital of Yunnan Province (Kunhua Hospital/The Affiliated Hospital of Kunming University of Science and Technology), Kunming, China
- 2Translational Neurosurgery and Neurobiology, University Hospital Aachen, RWTH Aachen, Aachen, Germany
Mitochondrial autophagy is an early defense and protection process that selectively clears dysfunctional or excessive mitochondria through a distinctive mechanism to maintain intracellular homeostasis. Mitochondrial dysfunction during cerebral stroke involves metabolic disbalance, oxidative stress, apoptosis, endoplasmic reticulum stress, and abnormal mitochondrial autophagy. This article reviews the research progress on the mechanism of mitochondrial autophagy in ischemic stroke to provide a theoretical basis for further research on mitochondrial autophagy and the treatment of ischemic stroke.
Introduction
Stroke is a sudden disease with disturbance of cerebral blood circulation, which has surpassed ischemic heart disease, lung cancer, chronic obstructive pulmonary disease, and liver cancer to become the disease with the highest age-standardized mortality rate in China. According to the latest disease burden report in 2015, stroke has become not only the first cause of disease burden, but also the second leading cause of death and disability in the world (GBD 2015 DALYs and HALE Collaborators, 2016; GBD 2015 Mortality and Causes of Death Collaborators, 2016), and an important cause of premature death. Stroke is primarily divided into two categories: ischemic stroke (accounting for 60% of all strokes) and hemorrhagic stroke (Krishnamurthi et al., 2013). At present, studies have found a substantial link between mitochondrial dysfunction and the occurrence and development of ischemic stroke. Moreover, the abnormal autophagy is profoundly involved in mitochondrial dysfunction.
Autophagy
Autophagy is a process defined by the degradation of abnormal proteins and damaged organelles by bilayer membranes such as exfoliated endoplasmic reticulum or Golgi complex under physiological or pathological circumstances and mediated by autophagy-related proteins. The bilayer structures fuse with lysosomes to form autophagy lysosomes, which degrades the contents of biological macromolecules and organelles, so as to maintain cell homeostasis by meeting the needs of cellular metabolism and organelle renewal (Parzych and Klionsky, 2014; Tagaya and Arasaki, 2017). Autophagy is essential for neuronal survival in the central nervous system. According to the mechanism and the mode of action, autophagy can be divided into three types: macroautophagy, microautophagy, and chaperone-mediated autophagy (CMA). Among them, macroautophagy is the most thoroughly studied. Mitochondrial autophagy, also called mitophagy, is a kind of macrophage autophagy, which selectively degrades damaged mitochondria through autophagy mechanism to sustain the stability of the intracellular environment. Mitochondrial autophagy plays an important role in regulating cellular homeostasis, proliferation, differentiation, genetics, aging, and cell death, as well as in controlling the quantity and quality of mitochondria (Glick et al., 2010).
The Regulatory Mechanism of Mitochondrial Autophagy
PTEN Induced Putative Kinase 1 (PINK1)/Parkin Signaling in Mitochondrial Autophagy
PTEN Induced Putative Kinase 1 (PINK1)/Parkin-mediated autophagy is the most common type of mitochondrial autophagy in mammalian cells (He et al., 2019). PINK1 is a nuclear-encoded mitochondrial serine (Ser)/threonine kinase (Eiyama and Okamoto, 2015; Quinn et al., 2020), which is divided into N-terminal mitochondrial targeting signal (MTS), α-helical transmembrane domain (TM), serine/threonine kinase domain, and C-terminal mitochondrial outer membrane retention signal peptide sequence (Deas et al., 2011), it can mediate the ubiquitination of substrates, and can regulate protein degradation and signal transduction. Parkin is an E3 ubiquitin ligase encoded by the Park2 gene. Genetic studies have shown that PINK1 plays a role in the upstream of Parkin, and PINK1 and Parkin are in the same pathway to preserve mitochondrial functions (Poole et al., 2008; Whitworth and Pallanck, 2009; Tanaka, 2020). In healthy cells, PINK1 is transported to the mitochondrial inner membrane, where it is cut and degraded rapidly by the ubiquitin-protease system, when the intracellular mitochondrial membrane is damaged, and consequently, the transmembrane potential is disturbed, the transport of PINK1 into the mitochondria will be blocked and PINK1 will accumulate in the outer membrane of the mitochondria, which then mediates the activation of Parkin and ubiquitin phosphorylation (Poole et al., 2008; Jin and Youle, 2013; Durcan and Fon, 2015; Okatsu et al., 2015a; Yamano et al., 2016). PINK1 can simultaneously phosphorylate Ser65 in the N-terminal ubiquitin-like domain of Parkin and produce Ser65 phosphorylated Parkin (Kondapalli et al., 2012; Iguchi et al., 2013; Okatsu et al., 2015b; Yang et al., 2020), phosphorylated Ser65 activates its ubiquitin ligase activity, which is able to connect the polyubiquitin chain to the mitochondrial outer membrane protein (Kazlauskaite et al., 2014), the complex is recognized by LC3, binds to the autophagosome, and then fuses with the lysosome to form the autophagy lysosome that finally degrades the damaged mitochondria. Studies from the Boule laboratory have confirmed that PINK1/Parkin-regulated mitochondrial autophagy plays a key role in the clearance of damaged mitochondria (Lazarou et al., 2015). Mitochondria maintain metabolic balance through continuous fusion and division. Additionally, the fusion/division and the mitochondrial autophagy influence each other; malfunctioning mitochondria are effectively removed by mitochondrial autophagy, and only the mitochondria with normal membrane potential can enter the normal fusion-division cycle (Twig et al., 2008), PINK1 and Parkin are of importance in the process of mitochondrial fusion and division. Related studies have shown that PINK1 and Parkin can promote mitochondrial division and regulate mitochondrial movement by mediating Miro phosphorylation to isolate damaged mitochondria (Poole et al., 2008), maintaining energy balance, and avoiding oxidative stress (Zhuang et al., 2016; Figure 1).
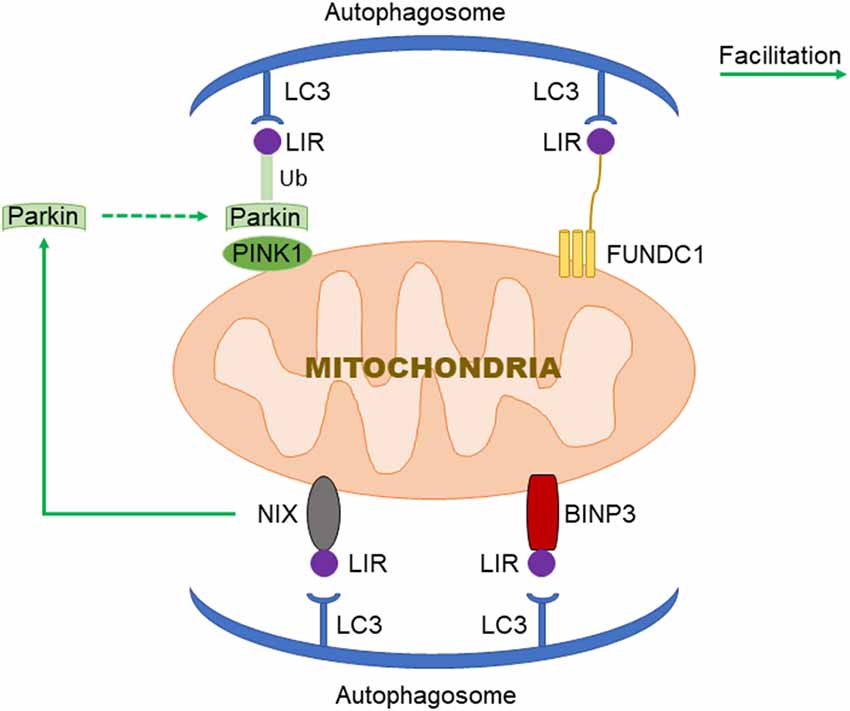
Figure 1. Three signaling pathways regulating mitochondrial autophagy. In PTEN Induced Putative Kinase 1 (PINK1)/Parkin pathway, PINK1 functions as the initiator which recognizes the disturbed outer membrane potential and activates Parkin and ubiquitin phosphorylation, and then the complex is combined by LC3 to autophagosome; FUN14 Domain-Containing 1 (FUNDC1) is a three transmembrane protein having the LC3 interaction region (LIR) motif on the N-terminal which can be recognized by LC3; both BNIP3 and NIX have LIR motif, additionally, NIX can facilitate the localization of Parkin to the damaged mitochondria.
Mitochondrial Autophagy Regulated by FUN14 Domain-Containing 1 (FUNDC1)
FUN14 Domain-Containing 1 (FUNDC1) is a new type of mitochondrial membrane protein that mediates mitochondrial autophagy in mammalian cells, it mainly acts in controlling mitochondrial autophagy by regulating the phosphorylation level of FUNDC1 on the mitochondrial membrane. FUNDC1 is a protein located in the outer membrane of mitochondria, which contains three transmembrane domains, as well as the N-terminal domain exposed to the cytoplasm and the C-terminal domain inserted into the mitochondrial inner membrane. It has been found that FUNDC1 contains a typical LC3 interaction region (LIR) sequence consisting of amino acid residues at the N-terminal on the cytoplasmic side (Liu et al., 2014; Cai et al., 2021). Under physiological conditions, FUNDC1 inhibits mitochondrial autophagy due to the phosphorylation of Tyr18 in the LIR motif of sarcoma (Src) gene kinase, leading to the inability of FUNDC1 to recruit LC3 and to combine with it, which reduces the interaction between FUNDC1 and LC3 (Liu et al., 2012; Chen et al., 2016; Kuang et al., 2016). Studies have shown that mitochondrial autophagy receptor protein, FUNDC1, is involved in mitochondrial autophagy under the condition of hypoxia in mammalian cells (Wang et al., 2018), when Src kinase and CK2 (formerly known as casein kinase 2) are inactivated during hypoxia or mitochondrial uncoupling, Ser17 on FUNDC1 is dephosphorylated by unc-51-like kinase 1 (ULK1) and Ser13 is dephosphorylated by phosphoglycerol mutase family 5 (PGAM5; Liu et al., 2014; Chen et al., 2016, 2017; Kuang et al., 2016), enhancing FUNDC-LC3 interaction and mitochondrial autophagy (Imai et al., 2010; Liu et al., 2014; Lv et al., 2017). Thus, it can be seen that FUNDC1 phosphorylation or dephosphorylation functions as a specific mitochondrial autophagy receptor in the process of mitochondrial autophagy induced by hypoxia. In addition, FUNDC1 can regulate mitochondrial division-fusion and mitochondrial autophagy by interacting with mitochondrial mitotic protein, dynamin-related protein 1 (Drp1), and optic nerve dystrophin 1 or with Drp1 and calcitonin (Ding and Yin, 2012; Iguchi et al., 2013; Wu et al., 2016a).
BNIP3/NIX Pathway in Mitochondrial Autophagy
BNIP3 (B Lymphoma-2 gene/adenovirus E1B interacting protein 3) and NIX (BNIP3L) are pro-apoptotic mitochondrial proteins located in the outer membrane of mitochondria (Kubli et al., 2007; Liu et al., 2019; Xu et al., 2019), they have 56% homology in amino acid sequence. Under certain conditions, the LIR sequence interacts with the recruited autophagy related protein, LC3, to form an autophagosome to clear the damaged mitochondria (Springer and Macleod, 2016; Šprung et al., 2018; Roperto et al., 2019). It has been reported that BNIP3/NIX mediates mitochondrial autophagy through upstream hypoxia-inducible 1 factor (HIF-1) regulation (Chourasia and Macleod, 2015); the competitive binding of BNIP3 and NIX to the anti-apoptotic protein, Bcl-2, will make Bcl-2-Beclin-1 complex dissociate and release Beclin-1, subsequently activating mitochondrial autophagy and reducing the apoptosis (Zhang et al., 2008). BNIP3 and NIX can promote the enhancement of mitochondrial autophagy by inhibiting the mammalian rapamycin target protein (mTOR) to activate protein Rheb. The upregulation of BNIP3 and NIX expression can be mediated by forkhead box O3 (FOXO3). Some studies have found that BNIP3 and NIX can not only induce cell death, but also increase their expression level under hypoxia, reducing reactive oxygen species (ROS) production and promoting cell survival, and can participate in the regulation of mitochondrial autophagy (de Vries and Przedborski, 2013). BNIP3 can increase the localization of mitochondrial kinesin-associated protein 1, thus promoting the phagocytosis of damaged mitochondria (Liu, 2015). Additionally, BNIP3 promotes the release of cytochrome C. Other studies have found that mitochondrial oxidative phosphorylation uncoupling agent carbonyl cyanide m-chlorophenyl hydrazone (CCCP) treatment on Hela cells can improve the interaction between NIX and LC3, indicating that NIX may also be involved in depolarization-induced mitochondrial autophagy (Novak et al., 2010). NIX mainly regulates mitochondrial autophagy at baseline under physiological conditions and mediates mitochondrial clearance during erythrocyte development by increasing the production of ROS (Novak and Dikic, 2011; Shi et al., 2014). Moreover, NIX can promote the localization of Parkin to the damaged mitochondria, increase the ubiquitination of mitochondrial membrane proteins by Parkin, and eventually induce mitochondrial autophagy through the interaction between p62 and LC3 (Geisler et al., 2010).
Mitochondrial Autophagy and Stroke
Neurological impairment caused by mitochondrial dysfunction is significantly associated with stroke, the main mechanisms include increased adenosine triphosphate (ATP), increased ROS level, and oxidative stress overload. Under mild ischemia or tolerable hypoxic stress, mitochondrial autophagy can benefit cell homeostasis and promote cell survival. On the contrary, vulnerable brain cells in the ischemic core area of persistent ischemia or reperfusion begin to die, which will lead to excessive long–term autophagy hyperactivity, causing cell damage or death (Zhang X. et al., 2013). Therefore, in the course of a stroke, it may be a possible solution of protecting neurons and preventing cell death to regulate the quantity and quality of mitochondria through the process of mitochondrial autophagy (Wong and Cuervo, 2010; Figure 2).
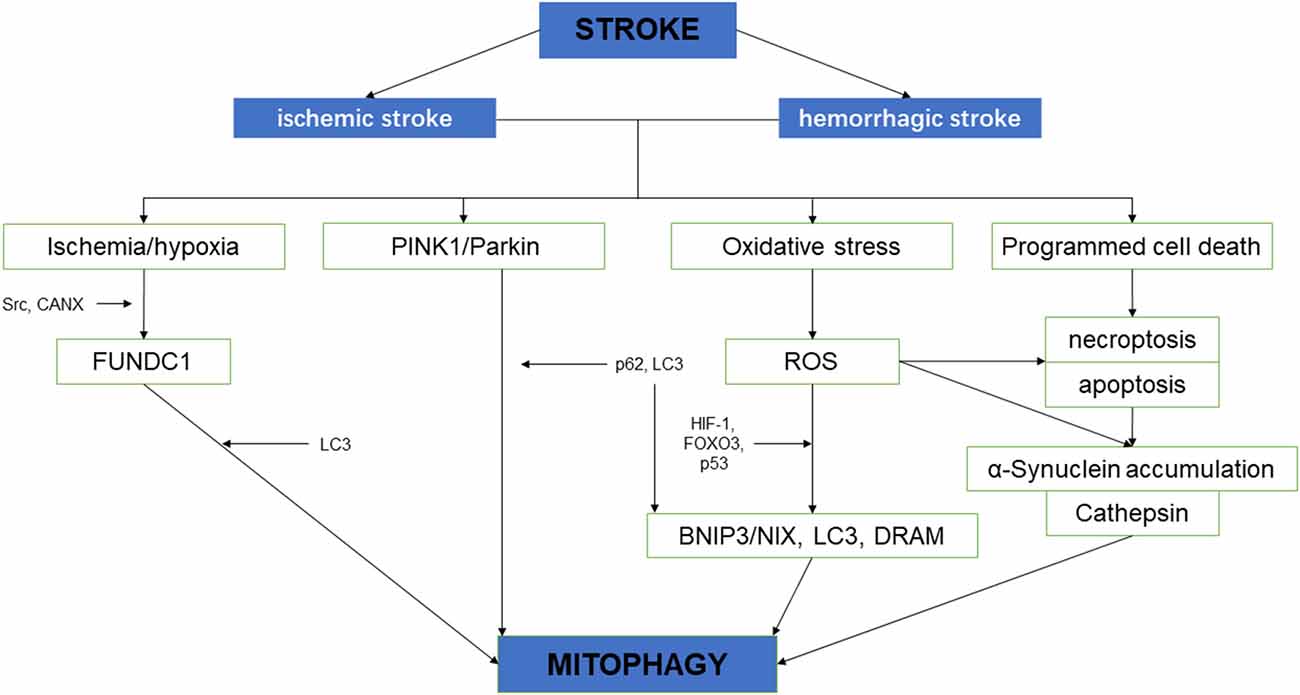
Figure 2. The schematic diagram depicting the relationship between cerebral stroke and mitochondrial autophagy.
No comments:
Post a Comment